Aktiva ingredienser: Epoetin zeta
Retacrit 1 000 IE / 0,3 ml injektionsvätska, lösning i en förfylld spruta
Retacrit 2000 IE / 0,6 ml injektionsvätska, lösning i en förfylld spruta
Retacrit 3000 IE / 0,9 ml injektionsvätska, lösning i förfylld spruta
Retacrit 4000 IE / 0,4 ml injektionsvätska, lösning i förfylld spruta
Retacrit 5 000 IE / 0,5 ml injektionsvätska, lösning i förfylld spruta
Retacrit 6000 IE / 0,6 ml injektionsvätska, lösning i en förfylld spruta
Retacrit 8 000 IE / 0,8 ml injektionsvätska, lösning i förfylld spruta
Retacrit 10 000 IE / 1 ml injektionsvätska, lösning i förfylld spruta
Retacrit 20 000 IE / 0,5 ml injektionsvätska, lösning i en förfylld spruta
Retacrit 30 000 IE / 0,75 ml injektionsvätska, lösning i en förfylld spruta
Retacrit 40 000 IE / 1 ml injektionsvätska, lösning i förfylld spruta
Indikationer Varför används Retacrit? Vad är det för?
Retacrit innehåller ett protein som kallas epoetin zeta som stimulerar benmärgen att göra fler röda blodkroppar i blodet som bär hemoglobin (ett ämne som binder syre). Epoetin zeta är en kopia av det humana proteinet erytropoietin och fungerar på samma sätt.
Retacrit används:
- hos vuxna, barn och ungdomar som genomgår hemodialys, för behandling av symptomatisk anemi (minskat antal röda blodkroppar) i samband med kronisk njursvikt (njursjukdom);
- hos vuxna patienter som genomgår peritonealdialys, för behandling av symptomatisk anemi i samband med kronisk njursvikt (njursjukdom);
- hos vuxna patienter med nedsatt njurfunktion som ännu inte genomgår dialys, för behandling av svår anemi i samband med njursjukdom åtföljd av kliniska symptom;
- hos vuxna patienter som får kemoterapi för solida tumörer, malignt lymfom (cancer i lymfsystemet) eller multipelt myelom (cancer i benmärgen) för att behandla anemi och minska behovet av blodtransfusioner, om läkaren fastställer förekomsten av hög risk för behöver transfusioner; - hos patienter med måttlig anemi som är kandidater för kirurgi för att donera blod före operationen, så att de kan få sitt eget blod under eller efter operationen (autolog predonation);
- hos måttligt anemiska vuxna patienter som planeras för större ortopedisk (ben) kirurgi (t.ex. höft- eller knäbytesbehandling) för att minska behovet av blodtransfusioner.
Kontraindikationer När Retacrit inte ska användas
Använd inte Retacrit:
- om du är allergisk mot erytropoietiner eller något annat innehållsämne i detta läkemedel (anges i avsnitt 6)
- om du har utvecklat en sjukdom som kallas 'Pure Red Cell Aplasia' (PRCA) efter behandling med någon typ av erytropoietin
- om du har högt blodtryck som inte kan kontrolleras tillräckligt med specifika läkemedel som sänker blodtrycket
- om du inte kan ta mediciner för att förtunna ditt blod
- om du donerar blod före operationen och:
- har haft hjärtinfarkt eller stroke under månaden före behandlingen
- lider av instabil angina pectoris (nyligen eller ökande bröstsmärta)
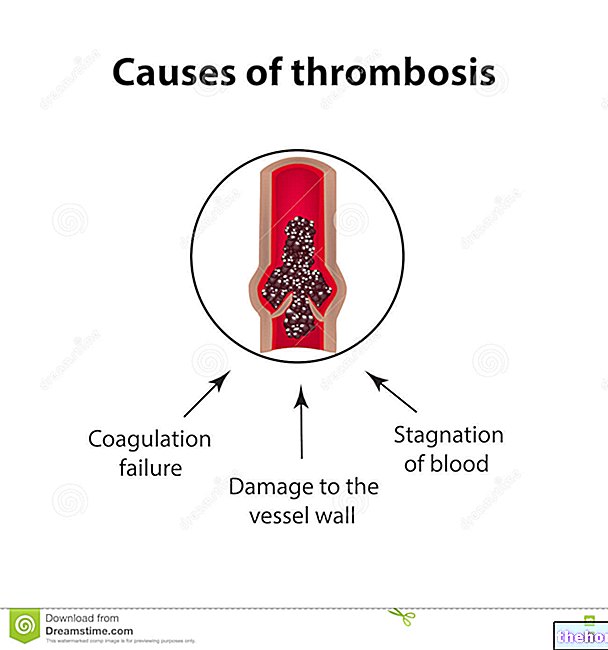
- riskerar att blodproppar bildas i venerna (djup venetrombos); till exempel om du tidigare har drabbats av trombos.
- Om du är på väg att genomgå en större ortopedisk operation, såsom höft- eller knäbyte, och:
- har allvarliga hjärt- eller blodcirkulationsproblem i vener eller artärer
- nyligen haft hjärtinfarkt eller stroke.
Försiktighetsåtgärder för användning Vad du behöver veta innan du tar Retacrit
Innan du använder Retacrit, tala om för din läkare om du vet att du har lidit eller lider av någon av följande sjukdomar:
- Beslag
- leversjukdom
- tumörer
- anemi på grund av andra orsaker
- hjärtsjukdom (såsom angina pectoris)
- blodcirkulationsstörningar som orsakar stickningar i extremiteterna, kalla händer eller fötter eller muskelkramper i benen
- trombos eller koagulationssjukdomar
- njursjukdom.
Interaktioner Vilka läkemedel eller livsmedel kan förändra effekten av Retacrit
Tala om för din läkare eller apotekspersonal om du tar eller nyligen har tagit eller kan tänkas ta andra läkemedel.
I synnerhet om du tar ett läkemedel som innehåller den aktiva substansen ciklosporin, för att hämma immunsystemet efter en njurtransplantation, kan din läkare beställa specifika test för att mäta koncentrationen av cyklosporin i blodet under behandling med Retacrit.
Att ta järntillskott och andra blodstimulantia kan öka effektiviteten av Retacrit. Din läkare kommer att avgöra om du ska fortsätta att ta dessa ämnen eller inte.
Varningar Det är viktigt att veta att:
Under behandling med Retacrit
Din läkare kommer att kontrollera att ditt hemoglobin inte överskrider en viss nivå, eftersom höga koncentrationer av hemoglobin kan utgöra en risk för hjärtats eller blodkärlens hälsa och kan öka risken för hjärtinfarkt, stroke och död.
Läkare bör försöka hålla hemoglobinnivåerna mellan 10 och 12 g / dl. Hemoglobinnivåerna bör inte överstiga 12 g / dl.
Din läkare kommer att kontrollera ditt blodtryck regelbundet medan du använder Retacrit. Om du upplever huvudvärk, särskilt plötslig, dunkande migrän, eller om du börjar känna dig förvirrad eller får anfall, tala omedelbart med din läkare eller sjuksköterska.
Dessa symptom kan i själva verket vara varningstecknen på en plötslig ökning av blodtrycket, en situation som skulle kräva akut terapeutiskt ingripande.
En ökning av blodplättarnas nivå (celler som bidrar till blodproppar) kan uppstå under behandling med detta läkemedel. Detta fenomen bör förbättras under behandlingen. Vi rekommenderar att du kontrollerar antalet blodplättar regelbundet under de första 8 veckorna av behandlingen.
Om du genomgår en läkarundersökning på ett sjukhus eller en privat klinik eller tar blodprov, kom ihåg att informera din läkare om den Retacrit -behandling du följer, eftersom detta läkemedel kan förändra ditt tillstånd och testresultat.
Var särskilt uppmärksam på andra produkter som stimulerar produktionen av röda blodkroppar:
Retacrit är en av en produktgrupp som stimulerar produktionen av röda blodkroppar liksom det humana proteinet erytropoietin. Vårdpersonal kommer alltid att registrera det exakta namnet på den produkt de använder.
Patienter med njursjukdom
Sällsynta fall av Pure Red Cell Aplasia (PRCA) har rapporterats efter månader eller år av behandling med andra erytropoietininnehållande läkemedel; denna möjlighet kan inte uteslutas med Retacrit.
Specifika röda blodplättar innebär att benmärgen inte kan producera tillräckligt med röda blodkroppar. I detta fall kan ett allvarligt tillstånd av anemi uppstå, vars symptom är: ovanlig trötthet, yrsel eller andnöd. Röd cellplasi kan orsakas av produktion av antikroppar riktade mot det injicerade erytropoietinet och därefter mot erytropoietinet som produceras av samma organism.
Diskutera denna information med din läkare. Om denna aplasi, dock sällsynt sjukdom, skulle inträffa, kommer behandlingen med Retacrit att avbrytas och läkaren beslutar vad man ska göra för att behandla anemi på det mest effektiva sättet. Du bör veta att om denna komplikation, hur sällsynt som helst, skulle inträffa, du måste sluta ta Retacrit och få regelbundna och eventuellt livslånga blodtransfusioner för behandling av anemi. Tala omedelbart för din läkare om du plötsligt känner dig väldigt trött eller andfådd.Din läkare kommer att avgöra hur effektivt Retacrit är för dig och avbryta behandlingen om det behövs.
Patienter med kronisk njursvikt som behandlas med erytropoietin måste genomgå tester med jämna mellanrum för att mäta nivån av hemoglobin (den del av de röda blodkropparna som bär syre) tills en konstant nivå uppnås, och därefter med jämna mellanrum, för att minimera risk för ökat blodtryck.
Om du har kronisk njursvikt, och i synnerhet om du inte svarar tillräckligt på Retacrit, kommer din läkare att kontrollera dosen Retacrit du får eftersom, om du inte svarar på behandlingen, kan upprepade doser av Retacrit öka risken för hjärta eller blodkärl och kan öka risken för hjärtinfarkt, stroke och död.
I isolerade fall har en ökning av kaliumnivåerna i blodet observerats. Hos patienter med kronisk njursvikt kan korrektion av anemi leda till en ökning av aptit och absorption av kalium och protein. Om du genomgår dialysbehandling när du börjar Retacrit -behandlingen kan du behöva justera dialysparametrar för att behålla urea, kreatinin- och kaliumnivåer inom önskat intervall och din läkare bestämmer.
Hos patienter med kronisk njursvikt bör serumelektrolyter (ämnen som finns i blodet) övervakas. Om serumkaliumvärdena är höga (eller ökar) bör man överväga att avbryta administreringen av Retacrit tills dessa värden har korrigerats.
Under behandling med Retacrit är det ofta nödvändigt under hemodialys att öka dosen heparin, ett särskilt blodförtunnande ämne, för att minimera risken för blodproppar.Om denna dos av heparin inte är optimal är det möjligt att en ocklusion inträffar i dialysatorn.
Cancerpatienter
Cancerpatienter är mer benägna att få trombos om de tar erytropoietinliknande läkemedel som Retacrit (se avsnitt 4.) Du bör därför tala med din läkare om fördelarna med Retacrit, särskilt om du är fet eller tidigare har haft problem med trombos eller blodproppar.
Cancerpatienter som behandlas med erytropoietin måste genomgå laboratorietester med jämna mellanrum för att mäta nivån av hemoglobin (den del av de röda blodkropparna som bär syre) tills en konstant nivå uppnås, och därefter med tidsfrister. Periodisk.
Om du har cancer bör du vara medveten om att Retacrit kan fungera som en tillväxtfaktor för blodkroppar och att det under vissa omständigheter kan ha negativa effekter på cancern. Beroende på den specifika situationen kan en blodtransfusion vara att föredra. Diskutera detta med din läkare.
Graviditet och amning
Om du är gravid eller ammar eller tror att du är gravid eller planerar att bli gravid, rådfråga din läkare eller apotekspersonal innan du tar detta läkemedel.
Om du är gravid eller ammar ska Retacrit endast användas om de potentiella fördelarna uppväger de potentiella riskerna för barnet.
Fråga din läkare om råd innan du tar något läkemedel.
Köra och använda maskiner
Retacrit har ingen eller mycket liten påverkan på förmågan att framföra fordon eller använda maskiner.
Retacrit innehåller fenylalanin
Detta läkemedel innehåller fenylalanin, ett ämne som kan vara farligt för personer med fenylketonuri (en enzymbrist av genetiskt ursprung som orsakar en ökad eliminering av ett kemiskt ämne (fenylketon) i urinen och kan orsaka störningar i nervsystemet).
Retacrit innehåller natrium
Detta läkemedel innehåller mindre än 1 mmol natrium (23 mg) per dos, vilket betyder att det anses vara '' natriumfritt ''.
Dosering och användningssätt Så här använder du Retacrit: Dosering
Retakritbehandling startas vanligtvis under medicinsk övervakning. Retacrit -injektioner kan ges av en läkare, sjuksköterska eller annan vårdpersonal.
Om Retacrit injiceras under huden (subkutant), efter att du har sett hur, kan du också injicera lösningen själv. Använd alltid detta läkemedel enligt läkarens anvisningar. Om du är osäker, tala med din läkare.
Dosinformation
Dosen baseras på kroppsvikt i kilogram. Din läkare kommer att beställa test, till exempel blodprov, för att avgöra om du behöver ta Retacrit och kommer att bedöma den exakta dosen av Retacrit, hur länge du kommer att behöva genomgå behandlingen och hur läkemedlet kommer att ges. Dessa beslut beror på orsaken till anemi.Din läkare kommer att använda den lägsta effektiva dosen för att kontrollera symtomen på anemi. Om du inte svarar tillräckligt på Retacrit kommer din läkare att kontrollera dosen du får och informera dig om han ändrar den.
Du kan få järntillskott både före och under behandling med Retacrit, för ökad effektivitet av behandlingen.
Användning till patienter med njursjukdom
Retacrit måste administreras antingen under huden (subkutant) eller genom injektion antingen i en ven eller genom en kateter placerad i en ven.
Användning av Retacrit hos vuxna patienter som får hemodialys
Läkaren kommer att hålla hemoglobinkoncentrationen mellan 10 och 12 g / dl (6,2 - 7,5 mmol / l).
Retacrit kan administreras under dialyspasset eller i slutet av sessionen.
Den rekommenderade startdosen är 50 IE / kg (internationella enheter per kilogram), ges 3 gånger per vecka. Om lösningen administreras i en ven bör den injiceras under 1 till 5 minuter.
Beroende på hur din anemi reagerar på behandlingen kan denna dos justeras ungefär var fjärde vecka tills situationen är under kontroll.Din läkare kommer att ordinera blodprov som ska utföras regelbundet för att säkerställa att läkemedlet fortsätter att ha önskad effekt. När situationen är under kontroll fortsätter du att ta Retacrit i regelbundna doser 2 eller 3 gånger per vecka. Dessa doser kanske inte är lika höga som de som ursprungligen mottogs.
Användning av Retacrit till barn och ungdomar
(≤18 år) som behandlas med hemodialys Hos barn kommer läkaren att hålla hemoglobinkoncentrationen mellan 9,5 och 11 g / dl.
Retacrit ska ges till patienten i slutet av dialyspasset.
Dosering för barn och ungdomar baseras på kroppsvikt i kilogram. Den rekommenderade startdosen är 50 IE / kg, ges 3 gånger per vecka genom injektion i en ven (under en varaktighet på 1-5 minuter).
Beroende på hur anemi reagerar på behandlingen kan denna dos justeras ungefär var fjärde vecka tills situationen är under kontroll.Din läkare kommer att beställa blodprov med jämna mellanrum för att säkerställa att detta händer.
Användning av Retacrit hos vuxna patienter som genomgår peritonealdialys
Läkaren kommer att hålla hemoglobinkoncentrationen mellan 10 och 12 g / dl.
Den rekommenderade startdosen är 50 IE / kg, som ska administreras två gånger i veckan.
Beroende på hur anemi reagerar på behandlingen kan denna dos justeras ungefär var fjärde vecka tills situationen är under kontroll.
Din läkare kommer att beställa blodprov med jämna mellanrum för att säkerställa att läkemedlet fortsätter att ha önskad effekt.
Användning av Retacrit hos vuxna patienter med njursjukdom men som inte genomgår dialys
Den rekommenderade startdosen är 50 IE / kg, ges 3 gånger per vecka.
Denna startdos kan justeras av din läkare tills situationen är under kontroll. När situationen är under kontroll fortsätter du att ta Retacrit i regelbundna doser (3 gånger i veckan, eller om det ges under huden (subkutant) kan det också ges en gång i veckan eller varannan vecka). Maximal dos bör inte överstiga 150 IE / kg 3 gånger i veckan, 240 IE / kg (upp till högst 20 000 IE) en gång i veckan eller 480 IE / kg (upp till högst 40 000 IE) en gång i veckan. varannan vecka.
Din läkare kommer att beställa blodprov för dig att göra regelbundet för att säkerställa att läkemedlet fortsätter att ha önskad effekt.
Om du behandlas med längre dosintervaller (mer än en gång i veckan) kanske du inte kan behålla dina Hb -nivåer på ett adekvat sätt och du kan behöva öka din dos Retacrit eller dess administreringsfrekvens.
Användning av Retacrit hos vuxna patienter som får kemoterapi
Din läkare kan börja Retacrit om din hemoglobinnivå är 10 g / dl eller mindre.
Efter behandlingens början kommer läkaren att hålla hemoglobinkoncentrationen mellan 10 och 12 g / dl.
Den rekommenderade startdosen är 150 IE / kg, som ska administreras 3 gånger per vecka genom subkutan injektion. Alternativt kan din läkare rekommendera en startdos på 450 IE / kg en gång i veckan. Din läkare kan justera din startdos baserat på ditt anemi -svar på behandlingen; du kommer att fortsätta att ta Retacrit i 1 månad efter avslutad kemoterapi.
Användning hos vuxna patienter som deltar i ett autologt predonationsprogram
Den rekommenderade startdosen är 600 IE / kg, ges två gånger i veckan genom injektion i en ven. Retacrit kommer att ges till dig under de 3 veckorna före operationen. Du kommer också att ta järntillskott före och under behandlingen med Retacrit för att öka effektiviteten av detta läkemedel.
Användning till vuxna patienter som planeras för större ortopedisk (ben) kirurgi
En dos på 600 IE / kg ges genom injektion under huden en gång i veckan i 3 veckor före operationen och på operationsdagen. I de fall då det är nödvändigt att minska tiden före interventionen administreras en dos på 300 IE / kg under de tio dagarna före interventionen, dagen för interventionen och under de fyra följande dagarna.Om blodprov före operation visar för höga hemoglobinnivåer avbryts behandlingen.
Det är också viktigt att järnhalten i blodet är normal under behandlingen med Retacrit. Vid behov kommer du att få järn i munnen dagligen, helst redan innan behandling med Retacrit påbörjas.
Information om administration
Den förfyllda sprutan Retacrit är klar att användas. Varje spruta är endast för en injektion. Retacrit injektionsvätska, lösning ska inte skakas eller blandas med andra lösningar.
Om Retacrit injiceras under huden bör den mängd som injiceras på ett enda ställe inte överstiga 1 ml. Övre lår och buk bort från naveln är bra injektionsställen. Byt injektionsstället varje dag.
När du använder Retacrit, följ alltid dessa instruktioner:
- Ta den förseglade blistret som innehåller sprutan och låt den nå rumstemperatur innan du använder den. För detta tar det 15 till 30 minuter.
- Ta bort sprutan från blisterkassan och kontrollera att lösningen är klar, färglös och praktiskt taget fri från synliga partiklar.
- Ta bort nålskyddet och släpp ut luften från nålen och sprutan genom att hålla sprutan upprätt och försiktigt trycka upp kolven.
- Injicera lösningen enligt instruktionerna från din läkare. Fråga din läkare eller apotekspersonal om något inte är klart för dig.
Använd inte Retacrit om:
- blåsan är öppen eller på annat sätt skadad;
- lösningen är inte färglös eller innehåller synliga partiklar i suspension;
- c "vätska har läckt ut från den förfyllda sprutan eller kondens är synlig inuti blistret som fortfarande är förseglat;
- du vet att läkemedlet har frysts av misstag eller tror att detta kan ha hänt.
Byt från intravenös till subkutan administrering
När situationen är under kontroll fortsätter du att ta Retacrit vid regelbundna doser. Din läkare kan bestämma att Retacrit ska ges genom injektion under huden (subkutant) snarare än i en ven (intravenöst).
När du byter från en administreringsmetod till den andra behöver inte dosen ändras, din läkare kan då beställa blodprov för att kontrollera om en dosjustering behövs eller inte.
Ge dig själv en injektion av Retacrit under huden
I början av behandlingen ges Retacrit vanligtvis av en läkare eller sjuksköterska. Därefter kan din läkare föreslå att du eller din vårdgivare lär sig att injicera under huden (subkutant).
- Försök inte injicera dig själv om din läkare eller sjuksköterska inte har berättat för dig hur.
- Använd alltid Retacrit enligt instruktion från din läkare eller sjuksköterska.
- Använd endast läkemedlet om det har förvarats korrekt (se avsnitt 5).
- Före användning, ta ut sprutan från kylskåpet och låt den nå rumstemperatur. Det tar vanligtvis 15-30 minuter.
Använd en enda dos Retacrit från varje spruta.
När läkemedlet administreras under huden (subkutant) är volymen normalt inte mer än 1 ml för en enda injektion.
Retacrit måste administreras ensamt och inte blandas med andra injektionsvätskor.
Skaka inte de förfyllda sprutorna. Långvarig och kraftig skakning kan skada medicinen. Använd inte läkemedlet om det har skakats kraftigt.
Hur du injicerar dig själv med förfyllda sprutor
- Ta ut sprutan från kylskåpet. Vätskan måste nå rumstemperatur. Ta inte bort sprutans nålskydd när det når rumstemperatur.
- Kontrollera sprutan för att se till att det är rätt dos, att den inte har gått ut, att den inte är skadad och att vätskan är klar och inte är frusen
- Välj injektionsstället. De mest lämpliga platserna för injektion är övre låret och buken, förutom området runt naveln. Byt injektionsstället varje gång.
- Tvätta händerna. Använd en antiseptisk torkduk för att desinficera injektionsstället.
- Håll sprutan i kroppen med den täckta nålen uppåt.
- Håll inte sprutan vid kolvhuvudet, kolven eller nålskyddet.
- Dra aldrig kolven mot dig.
- Ta inte bort nålskyddet på den förfyllda sprutan förrän du är redo att injicera Retacrit.
- Ta bort locket från sprutan genom att hålla fatet och dra försiktigt i locket utan att vrida det. Tryck inte på kolven, rör vid nålen eller skaka sprutan.
- Ta en hudveck mellan tummen och pekfingret utan att pressa den för mycket.
- Tryck in nålen hela vägen. Din läkare eller sjuksköterska kommer att ha visat dig hur.
- Tryck in kolven med tummen hela vägen in för att injicera hela mängden vätska. Skjut den långsamt och jämnt och håll huden nypad.
- När kolven trycks till dess gräns, dra ut nålen och släpp huden.
- När nålen tas bort från huden kan lite blod läcka från injektionsstället. Detta är normalt. Du kan desinficera injektionsstället genom att trycka på den antiseptiska torken några sekunder efter injektionen.
- Lägg den använda sprutan i en behållare för vass. Försök inte sätta tillbaka skyddskåpan på nålen.
- Kasta aldrig använda sprutor i behållare för hushållsavfall.
Användning av nålskyddsanordningen
Den förfyllda sprutan kan utrustas med en säkerhetsanordning för nålen som skyddar mot oavsiktlig nålstick.
- Utför injektionen enligt den teknik som beskrivs ovan.
- Medan du håller sprutan med fingrarna vilande på stödkanten, tryck på kolven tills injektionen av hela dosen är klar. Nålskyddssystemet aktiveras INTE om FULL dosen inte har administrerats.
- Ta bort nålen från huden, släpp sedan kolven och sprutan går framåt tills skölden har täckt nålen och snäpper på plats.
Om du har glömt att använda Retacrit
Använd inte en dubbel dos för att kompensera för en glömd tidigare dos.
Om du slutar att ta Retacrit
Avbryt inte behandlingen utan att först ha råd med din läkare.
Fråga din läkare om du har ytterligare frågor om användningen av Retacrit.
Överdosering Vad du ska göra om du har tagit för mycket Retacrit
Retacrit har en stor säkerhetsmarginal och biverkningar från en överdos av detta läkemedel är osannolika. Tala omedelbart för din läkare eller sjuksköterska om du tycker att du har injicerat för mycket Retacrit.
Biverkningar Vilka är biverkningarna av Retacrit
Liksom alla läkemedel kan detta läkemedel orsaka biverkningar men alla användare behöver inte få dem.
Tala omedelbart för din läkare om du har huvudvärk, särskilt om det är plötslig, skarp, migränliknande huvudvärk, om du känner dig förvirrad eller om du har anfall. Dessa symtom kan vara varningstecken på en plötslig ökning. Blodtryck, vilket kräver akut behandling.
Tala om för din läkare eller sjuksköterska om du märker någon av effekterna på denna lista.
Mycket vanliga biverkningar
Dessa kan drabba fler än 1 av 10 personer som behandlas med Retacrit.
- Influensaliknande symptom, huvudvärk, ledvärk, svaghetskänsla, trötthet och yrsel.
- Hos patienter med njursjukdom som ännu inte genomgår dialys har trängsel i luftvägarna, såsom täppt näsa och halsont, rapporterats.
Vanliga biverkningar
Dessa kan drabba upp till 1 av 10 personer som behandlas med Retacrit.
- Ökat blodtryck. Ökningen av blodtrycket kan kräva behandling med läkemedel (eller justering av de läkemedel du redan behandlas för ditt höga blodtryck). Din läkare kommer att kontrollera ditt blodtryck med jämna mellanrum under behandling med Retacrit, särskilt under behandling med Retacrit. " terapistart.
- Bröstsmärta, andfåddhet, smärtsam svullnad i benen som kan vara ett symptom på blodproppar (lungemboli, djup venetrombos).
- Stroke (otillräcklig blodtillförsel till hjärnan, vilket kan orsaka oförmåga att röra en eller flera lemmar på ena sidan av kroppen, oförmåga att förstå eller tala eller oförmåga att se ena sidan av synfältet).
- Utslag och svullnad runt ögonen (ödem), som kan orsakas av en allergisk reaktion.
- Koagulation i den konstgjorda njuren.
Mindre vanliga biverkningar
Dessa kan drabba upp till 1 av 100 personer som behandlas med Retacrit.
- Hjärnblödning.
Sällsynta biverkningar
Dessa kan drabba upp till 1 av 1 000 personer som behandlas med Retacrit.
- Överkänslighetsreaktioner.
Mycket sällsynta biverkningar
Dessa kan drabba upp till 1 av 10 000 personer som behandlas med Retacrit.
- Det kan finnas ökningar i blodplättarnas nivåer, som normalt är involverade i bildandet av blodproppar. Läkaren kommer att kontrollera dessa värden.
Biverkningar med frekvens inte känd
Frekvensen av dessa biverkningar kan inte beräknas utifrån tillgängliga data.
- Svullnad, särskilt i ögon- och läppområdet (Quinckes ödem) och chockliknande allergiska reaktioner med symtom som stickningar, rodnad, klåda, rodnad och snabb puls.
- Vaskulära och trombotiska händelser (blodproppar) i blodkärl såsom förhindrad blodtillförsel till hjärnan, retinal trombos, hindrad blodtillförsel till hjärtat, hjärtinfarkt, arteriell trombos, utvidgning av blodkärlens väggar (aneurysm).
- Red Series Aplasia (PRCA) PRCA har rapporterats hos patienter efter månader till år med subkutan (injektion under huden) behandling av erytropoietin. PRCA betyder oförmåga att producera ett tillräckligt antal röda blodkroppar i benmärgen (se avsnittet "Varningar och försiktighetsåtgärder").
- Klåda.
Andra biverkningar
Patienter med njursjukdom
- Ökning av blodtrycket, vilket kan kräva läkemedelsbehandling eller justering av dosen av läkemedel du redan tar för högt blodtryck. Din läkare kan mäta ditt blodtryck regelbundet medan du använder Retacrit, särskilt i början av behandlingen.
- En ocklusion av sambandet mellan artär och ven (shunttrombos) kan uppstå särskilt om du har lågt blodtryck eller om du har komplikationer av arteriovenös fistel.Din läkare kommer att kunna kontrollera shunten och ordinera ett läkemedel för att förhindra trombos.
Patienter med maligna tumörer
- Blodproppar (vaskulära trombotiska händelser) (se avsnittet "Varningar och försiktighetsåtgärder").
- Ökat blodtryck. Av denna anledning bör hemoglobinnivåer och blodtryck övervakas.
Tala om för din läkare, apotekspersonal eller sjuksköterska om du upplever några biverkningar. Detta gäller även biverkningar som inte anges i denna bipacksedel.
Rapportering av biverkningar
Tala med din läkare om du får några biverkningar. Detta inkluderar eventuella biverkningar som inte nämns i denna bipacksedel. Du kan också rapportera biverkningar direkt via det nationella rapporteringssystemet som anges i bilaga V. Genom att rapportera biverkningar kan du hjälpa till att ge mer information om läkemedlets säkerhet.
Giltighetstid och lagring
Förvara detta läkemedel utom syn- och räckhåll för barn.
Använd inte detta läkemedel efter utgångsdatum som anges på etiketten och kartongen ("EXP" / "EXP").
Utgångsdatumet avser den sista dagen i månaden.
Förvaras i kylskåp (2 ° C - 8 ° C). Frys inte.
Sprutan kan avlägsnas från kylskåpet och lämnas i rumstemperatur under en enda period på upp till 3 dagar (men inte över 25 ° C).
Förvara den förfyllda sprutan i ytterkartongen för att skydda läkemedlet från ljus.
Kasta inte några läkemedel via avloppsvatten eller hushållsavfall. Fråga din apotekare hur du ska kasta läkemedel som du inte längre använder. Detta hjälper till att skydda miljön.
Deadline "> Annan information
Vad Retacrit innehåller
Den aktiva ingrediensen är epoetin zeta (producerad med rekombinant DNA -teknik i äggstockcellinjer från kinesiska hamster).
Retacrit 1 000 IE / 0,3 ml injektionsvätska, lösning i en förfylld spruta
1 förfylld spruta med 0,3 ml injektionsvätska, lösning innehåller 1 000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 3333 IE epoetin zeta per ml.
Retacrit 2000 IE / 0,6 ml injektionsvätska, lösning i en förfylld spruta
1 förfylld spruta med 0,6 ml injektionsvätska, lösning innehåller 2000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 3333 IE epoetin zeta per ml.
Retacrit 3000 IE / 0,9 ml injektionsvätska, lösning i förfylld spruta
1 förfylld spruta med 0,9 ml injektionsvätska, lösning innehåller 3000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 3333 IE epoetin zeta per ml.
Retacrit 4000 IE / 0,4 ml injektionsvätska, lösning i förfylld spruta
1 förfylld spruta med 0,4 ml injektionsvätska, lösning innehåller 4000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 10 000 IE epoetin zeta per ml.
Retacrit 5 000 IE / 0,5 ml injektionsvätska, lösning i förfylld spruta
1 förfylld spruta med 0,5 ml injektionsvätska, lösning innehåller 5000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 10 000 IE epoetin zeta per ml.
Retacrit 6000 IE / 0,6 ml injektionsvätska, lösning i en förfylld spruta
1 förfylld spruta med 0,6 ml injektionsvätska, lösning innehåller 6000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 10 000 IE epoetin zeta per ml.
Retacrit 8 000 IE / 0,8 ml injektionsvätska, lösning i förfylld spruta
1 förfylld spruta med 0,8 ml injektionsvätska, lösning innehåller 8 000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 10 000 IE epoetin zeta per ml.
Retacrit 10 000 IE / 1 ml injektionsvätska, lösning i förfylld spruta
1 förfylld spruta med 1,0 ml injektionsvätska, lösning innehåller 10 000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 10 000 IE epoetin zeta per ml.
Retacrit 20 000 IE / 0,5 ml injektionsvätska, lösning i en förfylld spruta
1 förfylld spruta med 0,5 ml injektionsvätska, lösning innehåller 20 000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 40 000 IE epoetin zeta per ml.
Retacrit 30 000 IE / 0,75 ml injektionsvätska, lösning i en förfylld spruta
1 förfylld spruta med 0,75 ml injektionsvätska, lösning innehåller 30 000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 40 000 IE epoetin zeta per ml.
Retacrit 40 000 IE / 1 ml injektionsvätska, lösning i förfylld spruta
1 förfylld spruta med 1,0 ml injektionsvätska, lösning innehåller 40000 internationella enheter (IE) epoetin zeta (rekombinant humant erytropoietin). Lösningen innehåller 40 000 IE epoetin zeta per ml. Övriga innehållsämnen är dinatriumfosfatdihydrat, monobasiskt natriumfosfatdihydrat, natriumklorid, kalciumkloriddihydrat, polysorbat 20, glycin, leucin, isoleucin, treonin, glutaminsyra, fenylalanin och vatten för injektionsvätskor, natriumhydroxid (för pH -justering), saltsyra (för att justera pH).
Hur Retacrit ser ut och förpackningens innehåll
Retacrit är en klar och färglös injektionsvätska, lösning som finns i klara färglösa glassprutor med en fast nål.
De förfyllda sprutor innehåller 0,3 ml till 1 ml lösning, beroende på innehållet i epoetin zeta (se avsnitt "Innehåller Retacrit").
En förpackning innehåller 1 eller 4 eller 6 förfyllda sprutor med eller utan nålskydd.
Multipaket innehåller 4 (4 förpackningar om 1) eller 6 (6 förpackningar med 1) förfyllda sprutor.
Bipacksedel: AIFA (Italian Medicines Agency). Innehåll publicerat i januari 2016. Den information som finns finns kanske inte uppdaterad.
För att få tillgång till den senaste versionen är det lämpligt att gå till AIFA (Italian Medicines Agency) webbplats. Ansvarsfriskrivning och användbar information.
01.0 LÄKEMEDLETS NAMN -
RETACRIT 1000 IE / 0,3 ML LÖSNING FÖR INJEKTION I FÖRFYLLAD SPRUTA
02.0 KVALITATIV OCH KVANTITATIV SAMMANSÄTTNING -
En förfylld spruta med 0,3 ml injektionsvätska, lösning innehåller 1 000 internationella enheter (IE) epoetin zeta * (rekombinant humant erytropoietin). Lösningen innehåller 3333 IE epoetin zeta per ml.
* Producerat med rekombinant DNA -teknik i cellinjer från kinesisk hamster äggstock (CHO).
Hjälpämne med känd effekt:
Varje förfylld spruta innehåller 0,15 mg fenylalanin.
För fullständig förteckning över hjälpämnen, se avsnitt 6.1.
03.0 LÄKEMEDELSFORM -
Injektionsvätska, lösning i förfylld spruta.
Klar och färglös lösning.
04.0 KLINISK INFORMATION -
04.1 Terapeutiska indikationer -
• Behandling av symptomatisk anemi i samband med kronisk njursvikt (CRI) hos vuxna och barn:
• Behandling av anemi i samband med kronisk njursvikt hos vuxna och barn i hemodialys och hos vuxna patienter i peritonealdialys (se avsnitt 4.4).
• Behandling av svår anemi av njurursprung med kliniska symptom hos vuxna patienter med njurinsufficiens som ännu inte genomgår dialys (se avsnitt 4.4).
- Behandling av anemi och minskning av transfusionsbehov hos vuxna patienter som genomgår kemoterapi för solida tumörer, malignt lymfom eller multipelt myelom och riskerar blodtransfusion, vilket indikeras av patientens allmänna tillstånd (kardiovaskulär situation, anemi som redan existerade i början av kemoterapin) .
- Retacrit kan användas för att öka mängden autologt blod hos patienter som ingår i ett predonationsprogram. Användning i denna indikation bör utvärderas mot bakgrund av de rapporterade riskerna för tromboemboliska händelser.Behandling bör endast reserveras för patienter med måttlig anemi (i avsaknad av järnbrist) om blodkonserveringsförfaranden är otillgängliga eller otillräckliga när "Planerad större elektiv kirurgi kräver en stor volym blod (4 eller fler enheter blod för kvinnor, 5 eller fler enheter för män).
-Retacrit kan användas för att minska exponeringen för allogena blodtransfusioner hos icke -järnbristiga vuxna patienter som antas löpa stor risk för transfusionskomplikationer före större elektiv ortopedisk kirurgi. Begränsa användningen för patienter med måttlig anemi (Hb 10-13 g / dl ) ingår inte i ett autologt predonationsprogram och för vilket förväntas måttlig blodförlust (från 900 till 1 800 ml).
04.2 Dosering och administreringssätt -
Retakritterapi bör inledas under överinseende av medicinsk personal med erfarenhet av hantering av patienter med de indikationer som beskrivs ovan.
Dosering
Behandling av symptomatisk anemi hos vuxna och barn med kroniskt njursvikt
Retacrit ska administreras antingen subkutant eller intravenöst.
Den önskade hemoglobinkoncentrationen är mellan 10 och 12 g / dl (6,2-7,5 mmol / l), förutom hos barn, där hemoglobinkoncentrationen måste vara mellan 9,5 och 11 g / dl (5,9-6,8 mmol / l). Den övre gränsen för hemoglobinkoncentrationen bör inte överskridas. Symptom och följdsjukdomar för anemi kan variera med ålder, kön och övergripande sjukdomsbörda; det är nödvändigt att den kliniska kursen och tillståndet hos den enskilda patienten utvärderas av läkaren. Retacrit ska administreras antingen subkutant eller intravenöst för att uppnå hemoglobinvärden som inte överstiger 12 g / dl (7,5 mmol / l). På grund av variationen inom patienten kan enstaka hemoglobinvärden över och under den önskade hemoglobinkoncentrationen ibland observeras hos en patient. Variation i hemoglobin bör hanteras genom dosjustering, med hänvisning till ett målhemoglobinintervall mellan 10 g / dl (6,2 mmol / l) och 12 g / dl (7,5 mmol / l).
En förlängd hemoglobinnivå över 12 g / dl (7,5 mmol / l) bör undvikas; Anvisningar för lämplig dosjustering för när hemoglobinvärden över 12 g / dl (7,5 mmol / l) observeras ges nedan. En ökning av hemoglobin med mer än 2 g / dl (1,25 mmol / l) under en fyraveckorsperiod bör undvikas. Om detta inträffar bör lämplig dosjustering göras enligt anvisningarna.
Patienter bör övervakas noggrant för att säkerställa att den lägsta godkända effektiva dosen Retacrit används för att på ett adekvat sätt kontrollera symtomen på anemi genom att bibehålla en hemoglobinkoncentration på mindre än eller lika med 12 g / dl (7,5 mmol / l).
Försiktighet bör iakttas vid ökade doser av Retacrit hos patienter med kroniskt njursvikt.Patienter med dåligt hemoglobinsvar på Retacrit bör alternativa förklaringar till detta dåliga svar övervägas (se avsnitt 4.4 och 5.1) Hos patienter med kronisk nedsatt njurfunktion sjukdom och kliniska tecken på ischemisk hjärtsjukdom eller kongestivt hjärtsvikt, bör underhållshemoglobinkoncentrationen inte överskrida gränsvärdet för högsta målkoncentration.
Vuxna patienter i hemodialys
Retacrit ska administreras antingen subkutant eller intravenöst.
Behandlingen är indelad i två faser:
1. Korrigeringsfas: 50 IE / kg, 3 gånger i veckan. Om dosjustering är nödvändig bör detta göras gradvis, med minst 4 veckors intervall. För varje justering ska dosen ökas eller minskas med 25 IE / kg, 3 gånger per vecka.
2. Underhållsfas: Dosjustering som syftar till att upprätthålla önskad nivå av hemoglobin (Hb), mellan 10 och 12 g / dl (6,2-7,5 mmol / l). Den rekommenderade totala veckodosen varierar från 75 till 300 IE / kg.
Tillgängliga kliniska data indikerar att patienter med en mycket låg initial hemoglobinnivå (8 g / dl eller> 5 mmol / l).
Pediatriska patienter på hemodialys
Behandlingen är uppdelad i två faser.
1. Korrigeringsfas 50 IE / kg, 3 gånger per vecka intravenöst. Om dosjustering är nödvändig bör detta göras i steg om 25 IE / kg 3 gånger per vecka, med intervall om minst 4 veckor, tills målet uppnås.
2. Underhållsfas Dosjustering som syftar till att upprätthålla önskad nivå av hemoglobin (Hb), mellan 9,5 och 11 g / dl (5,9-6,8 mmol / l).
I allmänhet kräver barn och ungdomar som väger mindre än 30 kg högre underhållsdoser än barn som väger mer än 30 kg och vuxna. I kliniska studier observerades till exempel följande underhållsdoser efter 6 månaders behandling:
Tillgängliga kliniska data indikerar att patienter med en mycket låg initial hemoglobinnivå (6,8 g / dl eller> 4,25 mmol / l).
Vuxna patienter i peritonealdialys
Retacrit ska administreras antingen subkutant eller intravenöst.
Behandlingen är uppdelad i två faser.
1. Korrigeringsfas: Startdosen är 50 IE / kg vikt, två gånger i veckan.
2. Underhållsfas: Dosjustering som syftar till att bibehålla önskad nivå av hemoglobin (Hb), (mellan 10 och 12 g / dl [6,2-7,5 mmol / l]. Underhållsdosen är mellan 25 och 50 IE / kg 2 gånger per vecka, uppdelad i 2 lika stora doser.
Vuxna patienter med njurinsufficiens som ännu inte är i dialys
Retacrit ska administreras antingen subkutant eller intravenöst.
Behandlingen är uppdelad i två faser.
1. Korrigeringsfas: En initial dos på 50 IE / kg 3 gånger per vecka, följt vid behov av en ökning i steg om 25 IE / kg (3 gånger per vecka) tills önskat mål uppnås (ökningen bör ske gradvis, med minst fyra veckors intervall).
2. Underhållsfas: Under underhållsfasen kan Retacrit administreras 3 gånger per vecka och, vid subkutan administrering, en gång i veckan eller en gång varannan vecka. Dosen och doseringsintervallen måste justeras korrekt för att upprätthålla önskad hemoglobinnivå (Hb), (mellan 10 och 12 g / dl [6,2-7,5 mmol / l].
Förlängning av doseringsintervallet kan kräva en dosökning.
Maxdosen bör inte överstiga 150 IE / kg 3 gånger i veckan, 240 IE / kg (upp till högst 20000 IE) en gång i veckan eller 480 IE / kg (upp till högst 40 000 IE) en gång i veckan . varannan vecka.
Behandling av patienter med kemoterapiinducerad anemi
Retacrit ska ges subkutant till anemiska patienter (t.ex. med hemoglobinkoncentration ≤ 10 g / dl (6,2 mmol / l). Symptomen och konsekvenserna av anemi kan variera beroende på ålder, kön och svårighetsgrad. Övergripande sjukdom; en individuell bedömning av den kliniska kurs och tillstånd för varje enskild patient krävs av läkaren.
Med tanke på variationen inom patienten kan enstaka hemoglobinvärden över och under den önskade hemoglobinnivån ibland detekteras hos en patient. Variation i hemoglobin bör hanteras genom dosjustering, relativt ett målhemoglobinintervall på 10 g / dL (6,2 mmol / L) till 12 g / dL (7,5 mmol / L). En förlängd hemoglobinnivå över 12 g / dl (7,5 mmol / l) bör undvikas; Anvisningar för lämplig dosjustering för när hemoglobinvärden över 12 g / dl (7,5 mmol / l) observeras ges nedan.
Patienter bör övervakas noga för att säkerställa att den lägsta godkända dosen Retacrit används för att på ett adekvat sätt kontrollera symtomen på anemi.
Retakritterapi bör fortsätta i ytterligare en månad efter avslutad kemoterapi. Startdosen är 150 IE / kg, 3 gånger per vecka subkutant. Alternativt kan Retacrit administreras subkutant vid startdosen 450 IE / kg en gång i veckan. Om hemoglobinet efter 4 veckors behandling har ökat med minst 1 g / dL (0,62 mmol / L) eller om antalet retikulocyter har ökat med ≥ 40 000 celler / μl från baslinjen, bör dosen ligga kvar vid 450 IE / kg en gång i veckan eller 150 IE / kg 3 gånger i veckan. Om hemoglobinökningen är
Den rekommenderade doseringsregimen visas i följande tabell:
När det terapeutiska målet för den enskilda patienten har uppnåtts bör dosen minskas med 25 till 50% för att bibehålla hemoglobin på den nivån. Lämplig dostitrering bör övervägas.
Dosjustering
Om ökningen av hemoglobin är större än 2 g / dl (> 1,25 mmol / l) per månad bör dosen Retacrit minskas med cirka 25-50%. Om hemoglobinvärdet överstiger 12 g / dl (7,5 mmol / L ), avbryt behandlingen tills den återgår till eller sjunker under 12 g / dL (7,5 mmol / L), och fortsätt sedan Retacrit -behandlingen med en dos under 25% än den tidigare dosen.
Behandling av vuxna patienter som är kandidater för kirurgiska ingrepp som ingår i autologa predonationsprogram
Retacrit måste administreras intravenöst.
Vid tidpunkten för blodgivningen måste Retacrit administreras efter att donationsproceduren har slutförts.
Milt anemiska patienter (hematokrit 33-39%) som kräver en deposition på ≥ 4 enheter blod bör behandlas med 600 IE / kg Retacrit 2 gånger per vecka i 3 veckor före operationen.
Under hela behandlingen med Retacrit ska alla patienter få adekvat järntillskott (t.ex. 200 mg oralt elementärt järn per dag). Administreringen av järn bör påbörjas så snart som möjligt, till och med flera veckor innan autolog fördepåering utförs, för att öka järnlagren innan behandling med Retacrit påbörjas.
Behandling av vuxna patienter som planeras för större elektiv ortopedisk kirurgi
Retacrit ska administreras subkutant.
En dos på 600 IE / kg kroppsvikt bör administreras en gång i veckan i tre veckor (dag 21, 14 och 7) före operationen och på operationsdagen (dag 0). I de fall det är nödvändigt att minska tiden före operationen till mindre än tre veckor, bör en daglig dos på 300 IE / kg kroppsvikt administreras i 10 på varandra följande dagar före operationen, på operationsdagen och under de fyra dagarna om hemoglobinnivån når eller överstiger 15 g / dl som en del av hematologiska tester som utförts under den preoperativa perioden, Retacrit ska avbrytas och efterföljande doser ska inte ges.
Järnbrister måste behandlas innan behandling med Retacrit påbörjas. Dessutom bör en tillräcklig mängd järn administreras till alla patienter som behandlats med Retacrit (t.ex. 200 mg järnjoner oralt per dag) under hela behandlingsperioden med Retacrit.
Administreringssätt
Intravenös injektion
Administrering bör ta minst 1-5 minuter, beroende på den totala dosen. Hos hemodialyspatienter är det möjligt att administrera bolusdosen, under dialyspasset, från en lämplig venös åtkomst till dialyskretsen. Alternativt kan ämnet injiceras i slutet av dialyspasset genom fisteln och följt av 10 ml 9 mg / ml (0,9%) NaCl -fysiologisk lösning för att bevattna kretsen och säkerställa en tillfredsställande introduktion av produkten i cirkulationen. Hos patienter som reagerar på behandling med influensaliknande symptom är det att föredra att välja en långsammare administrering.
Retacrit får inte administreras genom intravenös infusion.
Retacrit får inte blandas med andra läkemedel (se avsnitt 6.2).
Subkutan injektion
I allmänhet bör den maximala volymen på 1 ml per injektionsställe inte överskridas.Vid större volymer är det nödvändigt att välja fler administreringsställen.
Injektioner ges i lemmarna eller i den främre bukväggen.
För instruktioner om hantering av läkemedlet före administrering, se avsnitt 6.6.
04.3 Kontraindikationer -
- Överkänslighet mot den aktiva substansen eller mot något hjälpämne som anges i avsnitt 6.1.
- Patienter med ren röd cellplasi (PRCA) efter behandling med erytropoietin ska inte genomgå behandling med Retacrit eller andra typer av erytropoietin (se avsnitt 4.4).
- Okontrollerad hypertoni.
- I indikationen "ökning av mängden autologt blod": hjärtinfarkt eller stroke under månaden före behandling, instabil angina pectoris, ökad risk för djup ventrombos, såsom en historia av tromboembolisk venös sjukdom.
- Vid indikation på större elektiv ortopedisk kirurgi: svår koronar, perifer arteriell, halspulsåder eller cerebral kärlsjukdom, inklusive patienter med nyligen hjärtinfarkt eller cerebrovaskulär olycka.
- Patienter som av någon anledning inte kan få "adekvat antitrombotisk profylax.
04.4 Varningar och lämpliga försiktighetsåtgärder vid användning -
Allmän information
Som med alla patienter som får erytropoietin kan en ökning av blodtrycket inträffa under behandling med Retacrit. Blodtrycket bör övervakas noggrant och kontrolleras på ett adekvat sätt före, vid start och under behandling med Retacrit, både hos alla patienter som behandlas med epoetin för första gången och hos patienter som redan behandlats. Det kan vara nödvändigt att etablera eller förstärka -hypertensiv behandling Om blodtrycket inte kan kontrolleras ska behandlingen med Retacrit avbrytas.
Retacrit ska också användas med försiktighet i närvaro av epilepsi och kroniskt leversvikt.
En måttlig dosberoende ökning av trombocytantalet inom det normala intervallet kan uppstå under behandling med erytropoietin. Detta fenomen går tillbaka med fortsatt behandling. Det rekommenderas att antalet trombocyter kontrolleras regelbundet under de första 8 veckorna av behandlingen.
Alla andra orsaker till anemi (järnbrist, hemolys, blodförlust, vitamin B12 eller folatbrist) bör utvärderas och behandlas före och under behandling med Retacrit. I de flesta fall minskar serumferritinvärdena samtidigt som hematokritvärdena ökar.För att säkerställa ett optimalt svar på erytropoietin måste tillräckliga järnlagrar säkerställas:
-för patienter med kronisk njurinsufficiens och serumferritinnivåer under 100 ng / ml järntillskott rekommenderas, till exempel 200-300 mg / dag oralt (100-200 mg / dag hos barn);
- Hos alla cancerpatienter med transferrinmättnadsvärden under 20%rekommenderas ett oralt järntillskott på 200-300 mg / dag.
Alla dessa faktorer som bidrar till uppkomsten av anemi måste också noga övervägas innan man bestämmer sig för att öka erytropoietindosen hos cancerpatienter.
En paradoxal minskning av hemoglobin och utveckling av allvarlig anemi i samband med lågt antal retikulocyter bör uppmärksamma dem på att avbryta behandling med epoetin och utföra antirytropoietinantikroppstest. Fall har rapporterats hos patienter med hepatit C som behandlats med interferon och ribavirin, när epoetiner. har använts i kombination. Epoetiner är inte godkända för behandling av anemi i samband med hepatit C.
För att förbättra spårbarheten för Erythropoiesis Stimulating Agents (Erytropiesstimulerande medel (ESA) måste namnet på den ESA som har förskrivits tydligt registreras (eller anges) i patientens journal.
God blodhanteringsmetod bör alltid användas vid perioperativ behandling.
Patienter planerade för större elektiv ortopedisk kirurgi
Hos patienter som planeras för större elektiv ortopedisk kirurgi bör orsakerna till anemi fastställas och behandlas, eventuellt innan behandling med Retacrit påbörjas. Trombotiska händelser kan utgöra en risk för denna patientpopulation, och detta bör noga övervägas i förhållande till den förväntade nyttan av behandlingen.Patienter bör få "adekvat antitrombotisk profylax, eftersom trombotiska och vaskulära händelser kan inträffa hos kirurgiska patienter, särskilt hos patienter med underliggande kardiovaskulär sjukdom. Dessutom bör särskild försiktighet iakttas hos patienter som är predisponerade för att utveckla djup venetrombos (DVT). Vidare bör patienter med hemoglobin> 13 g / dl vid baslinjen, kan möjligheten att Retacrit -behandling kan associeras med en ökad risk för postoperativa trombotiska / vaskulära händelser inte uteslutas.Därför bör sådan behandling inte användas till patienter med hemoglobin> 13 g / dl vid baslinjen .
Patienter med kronisk njurinsufficiens
Hemoglobinkoncentration
Hos patienter med kronisk njursvikt bör underhållskoncentrationen av hemoglobin inte överstiga den övre gränsen för den målhemoglobinkoncentration som rekommenderas i avsnitt 4.2. Ökad risk för död, allvarliga kardiovaskulära händelser och cerebrovaskulära händelser inklusive stroke har observerats i kliniska prövningar när ESA administrerades för att uppnå hemoglobinvärden större än 12 g / dl (7,5 mmol / l).
Kontrollerade kliniska prövningar har inte visat någon signifikant fördel som kan tillskrivas administrering av epoetiner när hemoglobinkoncentrationen har överskridit de nivåer som är nödvändiga för att kontrollera symtomen på anemi och undvika blodtransfusioner.
Hemoglobinnivån ska mätas med jämna mellanrum tills det når ett konstant värde, och därefter med periodiska intervall. Ökningen av hemoglobin bör vara cirka 1 g / dl (0,62 mmol / l) per månad och bör inte överstiga 2 g / dl (1,25 mmol / l) per månad för att minimera risken för att utveckla högt blodtryck eller förvärras.
Patienter med kronisk njursvikt som behandlas med Retacrit subkutant bör övervakas regelbundet med avseende på förlust av effekt, definierat som icke-svar eller minskat svar på Retacrit-behandling hos patienter som tidigare svarat på sådan behandling. Detta kännetecknas av en långvarig minskning av hemoglobin trots en ökning av dosen Retacrit.
Vissa patienter som behandlas med epoetin alfa med längre doseringsintervall (mer än en gång i veckan) kanske inte upprätthåller tillräckliga hemoglobinnivåer (se avsnitt 5.1) och kan kräva en dosökning. Hemoglobinnivåerna bör övervakas regelbundet.
Försiktighet bör iakttas vid ökande doser av Retacrit till patienter med kroniskt njursvikt eftersom höga kumulativa doser av epoetin kan vara associerade med en ökad risk för dödlighet och allvarliga kardiovaskulära och cerebrovaskulära händelser.Patienter med dåligt hemoglobinsvar på epoetiner, Alternativa förklaringar till detta dålig respons bör övervägas (se avsnitt 4.4 och 5.1).
Bristande respons på erytropoietinbehandling måste omedelbart undersöka de ansvariga faktorerna. Dessa inkluderar: järn-, folat- eller vitamin B12 -brist, aluminiumförgiftning, interkurrenta infektioner, inflammatoriska eller traumatiska episoder, ockult blodförlust, hemolys, benmärgsfibros av något ursprung.
Fall av antikroppsmedierat PRCA har mycket sällan rapporterats hos patienter med kroniskt njursvikt administrerat subkutant erytropoietin. Hos patienter som visar en "plötslig effektförlust, visad av en minskning av hemoglobin (1-2 g / dl per månad) med ökat behov av transfusioner, bör ett antal retikulocyter utföras och typiska orsaker som förhindrar respons på behandling (t.ex. järn , brist på folat eller vitamin B12, aluminiumförgiftning, infektion eller inflammation, blodförlust, hemolys). Om ingen orsak hittas bör man överväga att utföra ett blodprov benmärg för att diagnostisera en PRCA.
Om PRCA diagnostiseras ska Retacrit-behandlingen avbrytas omedelbart och ett test för förekomst av anti-erytropoietinantikroppar bör övervägas.Patienter ska inte omdirigeras till behandling med ett annat läkemedel, med korsreaktivitet mellan anti-erytropoietinantikroppar och andra erytropoietiner Andra orsaker till PRCA måste uteslutas och lämplig terapi sättas in.
Periodisk övervakning av antalet retikulocyter rekommenderas för att upptäcka eventuell förlust av terapeutisk effekt hos patienter med kroniskt njursvikt.
Hyperkalemi har observerats i isolerade fall. Hos patienter med kronisk njursvikt kan korrigering av anemi leda till ökad aptit och absorption av kalium och protein. Föreskrivna parametrar för dialys kan behöva periodisk justering för att hålla urea, kreatinin och kalium inom önskade värden. Hos patienter med kroniskt njursviktsserum elektrolyter bör övervakas. Om förhöjda (eller stigande) kaliumvärden i serum observeras bör man överväga att avbryta administrering av erytropoietin tills hyperkalemi har korrigerats.
En ökning av heparindosen krävs ofta under erytropoietinbehandling på grund av en ökning av hematokritvärdet. Ocklusion av dialyssystemet kan uppstå om heparinisering inte är optimal.
Baserat på tillgängliga data hittills, påskyndar inte korrigeringen av anemi med erytropoietin hos vuxna patienter med njurinsufficiens som ännu inte genomgår dialys utvecklingen av njurinsufficiens.
Vuxna cancerpatienter med symptomatisk anemi vid kemoterapi
Hos cancerpatienter som får kemoterapi bör intervallet 2-3 veckor mellan administrering och utseende av erytropoietininducerade erytrocyter beaktas vid bedömningen av lämpligheten av Retacrit-behandling (patienter med risk för transfusion). Hos cancerpatienter som genomgår kemoterapi, om hemoglobinet ökar mer än 2 g / dl (1,25 mmol / l) per månad eller om dess nivå överstiger 12 g / dl (7,5 mmol / l) är det Dosjusteringsförfarandet som anges i avsnitt 4.2 måste utföras noggrant för att minska potentialen riskfaktorer för trombotiska händelser (se avsnitt 4.2).
Eftersom en ökad förekomst av tromboemboliska händelser har observerats hos cancerpatienter som genomgår behandling med erytropoetiska medel (se avsnitt 4.8), bör denna risk vägas noggrant mot bakgrund av nyttan av behandlingen (med Retacrit), särskilt hos de cancerpatienter som presenterar en ökad tromboembolisk risk, såsom överviktiga personer eller som tidigare haft trombotiska och vaskulära händelser (djup venetrombos, lungemboli).
Vuxna patienter som är kandidater för kirurgi som ingår i ett autologt predonationsprogram
Alla varningar och speciella försiktighetsåtgärder i samband med autologa predonationsprogram måste följas, särskilt genom att återställa blodvolymen som vanligt.
Onkogen potential
Epoetiner är tillväxtfaktorer som främst stimulerar produktionen av erytrocyter. Erytropoietinreceptorer kan uttryckas på ytan av ett antal neoplastiska celler. Som med alla tillväxtfaktorer råder det tvivel om att epoetiner kan stimulera tillväxten av alla maligna tumörer. I flera kontrollerade kliniska prövningar har detta inte varit fallet. Epoetiner har har visat sig förbättra den övergripande överlevnaden eller minska risken för cancerprogression hos patienter med cancerassocierad anemi.
Flera kontrollerade kliniska prövningar där epoetiner administrerades till patienter med ett antal vanliga maligniteter, såsom skivepitelcancer i huvud- och halsområdet, lungcancer och bröstcancer, har visat en oförklarlig ökning av dödligheten. I kontrollerade kliniska prövningar visade användningen av epoetin alfa och andra erytropoiesstimulerande medel (ESA):
• En minskning av tiden till tumörprogression hos patienter med avancerad cancer i huvud och nacke som behandlats med strålbehandling vid administrering för att uppnå hemoglobinvärden större än 14 g / dl (8,7 mmol / l),
• en minskning av total överlevnad och en ökning av dödsfall som hänför sig till tumörprogression vid 4 månader hos patienter med metastaserad bröstcancer som behandlats med kemoterapi vid administrering för att uppnå hemoglobinvärden på 12-14 g / dl (7,5-8,7 mmol / l) ,
• ökad dödsrisk vid administrering för att erhålla hemoglobinvärden på 12 g / dl (7,5 mmol / l) hos patienter med aktiva maligniteter, som inte behandlas med kemoterapi eller strålbehandling. Användning av ESA är inte indicerat i denna patientpopulation.
Baserat på ovanstående, vid vissa kliniska tillstånd bör blodtransfusion vara den föredragna behandlingen för behandling av anemi hos cancerpatienter. Beslutet att administrera rekombinanta erytropoietiner bör baseras på en bedömning av nytta-risk-förhållandet med deltagande av den enskilda patienten och bör ta hänsyn till det specifika kliniska sammanhanget. Faktorer som ska beaktas i denna bedömning bör inkludera cancertyp och dess stadium, anemi, livslängd, miljö där patienten behandlas och patientens preferenser (se avsnitt 5.1).
Detta läkemedel innehåller fenylalanin, ett ämne som kan vara farligt för personer med fenylketonuri.
Detta läkemedel innehåller mindre än 1 mmol natrium (23 mg) per dos, vilket betyder att det anses vara "lågt i natrium".
04.5 Interaktioner med andra läkemedel och andra former av interaktion -
Behandling med erytropoietin har inte visat sig förändra metabolismen av andra läkemedel. Men eftersom cyklosporin binder till erytrocyter kan det finnas möjlighet till en "interaktion med andra läkemedel. Om erytropoietin administreras samtidigt med cyklosporin, bör blodnivåerna av cyklosporin övervakas och dosen av detta läkemedel måste korrigeras enligt ökningen av hematokritvärdet.
Det finns inga bevis som tyder på en "interaktion mellan epoetin alfa och G-CSF eller GM-CSF med avseende på hematologisk differentiering eller proliferation i tumörbiopsiprover. in vitro.
04.6 Graviditet och amning -
Det finns inga adekvata och välkontrollerade studier på gravida kvinnor. Djurstudier har visat reproduktionstoxicitet (se avsnitt 5.3). Det är inte känt om exogent epoetin zeta utsöndras i bröstmjölk. Därför ska erytropoietin i allmänhet endast användas under graviditet och amning om de potentiella fördelarna överväger de potentiella riskerna för fostret.
04.7 Effekter på förmågan att framföra fordon och använda maskiner -
Retacrit har ingen eller försumbar effekt på förmågan att framföra fordon eller använda maskiner.
04.8 Biverkningar -
Sammanfattning av säkerhetsprofilen
Resultaten av kliniska studier med Retacrit överensstämmer med säkerhetsprofilen för andra godkända erytropoietiner. Baserat på resultaten från kliniska studier med andra licensierade erytropoietiner förväntas cirka 8% av patienterna som behandlas med erytropoietin uppleva biverkningar Biverkningar under behandling med erytropoietin observeras främst hos patienter med kroniskt njursvikt eller underliggande maligniteter och de är huvudsakligen representerade genom huvudvärk och en dosberoende ökning av blodtrycket Hypertensiva kriser med symptom som liknar en "encefalopati" kan uppstå. Uppmärksamhet bör ägnas åt plötslig akut migränliknande huvudvärk, vilket kan vara ett varningstecken.
Andning i luftvägarna, inklusive händelser av överbelastning i övre delen, nästäppa och nasofaryngit, har rapporterats i vissa studier på vuxna patienter med nedsatt njurfunktion som ännu inte genomgått dialys som behandlats med längre doseringsintervall.
Hos patienter som behandlas med erytropoetiska medel har trombotiska / vaskulära händelser som myokardiskemi, hjärtinfarkt, cerebrovaskulära olyckor (hjärnblödning och hjärninfarkt), övergående ischemiska attacker, djup venetrombos, artär trombos, lungemboli, aneurysm, aneurysm, retinal trombos koagulation i den artificiella njuren.
Antikroppsmedierad erytroblastopeni (PRCA) har observerats efter månader eller år av behandling med epoetin alfa. Antikroppar mot erytropoietiner har observerats hos de flesta av dessa patienter (se avsnitt 4.3 och 4.4).
Utskrift av biverkningar
Detta avsnitt definierar frekvensen av biverkningar som: Mycket vanligt (> 1/10); vanligt (> 1/100 till 1/1 000 till 1/10 000 a
Inom varje frekvensklass presenteras biverkningar i fallande allvarlighetsordning.
Frekvensen kan variera beroende på indikationen
Vuxna och pediatriska hemodialyspatienter, vuxna patienter som genomgår peritonealdialys och vuxna patienter med nedsatt njurfunktion som ännu inte är i dialys
Den vanligaste biverkningen vid behandling med epoetin alfa är dosberoende höjning av blodtrycket eller försämring av redan existerande hypertoni. Denna ökning av blodtrycket kan behandlas farmakologiskt. Dessutom rekommenderas övervakning av blodtrycket. Särskilt i början av behandlingen. Följande reaktioner har också inträffat i isolerade fall av patienter med normalt eller lågt blodtryck: hypertensiv kris med symptom som liknar encefalopati (huvudvärk och förvirring) och generaliserade tonikokloniska anfall, som kräver omedelbar medicinsk intervention och intensiv behandling. Du bör vara särskilt uppmärksam till plötslig akut migränliknande huvudvärk, vilket kan vara ett varningstecken.
Shunttrombos kan förekomma, särskilt hos patienter med tendens till hypotoni eller med komplikationer av arteriovenösa fistlar (stenos, aneurysm etc.) Hos dessa patienter rekommenderas tidig revision av shunten och antitrombotisk profylax, till exempel med acetylsalicylsyra. .
Vuxna cancerpatienter på kemoterapi med symptomatisk anemi
Hypertoni kan förekomma hos patienter som behandlas med epoetin alfa. Följaktligen bör noggrann övervakning av hemoglobin och blodtryck utföras.
En ökad förekomst av vaskulära trombotiska händelser har observerats hos patienter som behandlats med erytropoetiska medel (se avsnitt 4.4 och avsnitt 4.8 - Allmänna överväganden).
Patientkandidater för operation
Oavsett behandling med erytropoietin kan tromboemboliska händelser inträffa efter upprepade flebotomier hos kirurgiska patienter med underliggande kardiovaskulär sjukdom.
Därför bör sådana patienter rutinmässigt genomgå ersättning av den uppsamlade blodvolymen. Hos patienter med hemoglobin> 13 g / dl vid baslinjen kan möjligheten att Retacrit -behandling kan associeras med en ökad risk för postoperativa trombotiska / vaskulära händelser inte uteslutas.
Rapportering av misstänkta biverkningar
Rapportering av misstänkta biverkningar som inträffar efter godkännande av läkemedlet är viktigt eftersom det möjliggör kontinuerlig övervakning av läkemedlets nytta / riskbalans.Hälso- och sjukvårdspersonal uppmanas att rapportera alla misstänkta biverkningar via det nationella rapporteringssystemet.
04.9 Överdosering -
Det terapeutiska fönstret för erytropoietin är mycket brett. Överdosering av erytropoietin kan ge effekter som är förlängningar av hormonets farmakologiska effekter. Om alltför höga hemoglobinnivåer inträffar kan en flebotomi utföras. Vid behov bör ytterligare stödjande vård ges.
05.0 FARMAKOLOGISKA EGENSKAPER -
05.1 "Farmakodynamiska egenskaper -
Farmakoterapeutisk grupp: Andra antianemika, erytropoietin
ATC -kod: B03XA01
Retacrit är ett biosimilärt läkemedel. Detaljerad information finns tillgänglig på Europeiska läkemedelsmyndighetens webbplats http://www.ema.europa.eu
Farmakodynamiska effekter
Erytropoietin är ett glykoprotein som, som en mitosstimulerande faktor och differentieringshormon, stimulerar produktionen av erytrocyter från stamfackets föregångare.Erytropoietins skenbara molekylvikt är 32.000-40.000 dalton. Proteinfraktionen av molekylen utgör cirka 58% av dess totala molekylvikt och består av 165 aminosyror. De fyra kolhydratkedjorna är kopplade till proteinet genom tre N-glykosidbindningar och en oglykosidbindning. Ur aminosyrasekvensens och kolhydratkompositionens synvinkel är epoetin zeta identiskt med endogent humant erytropoietin isolerat från urin från anemiska patienter. Den biologiska effekten av erytropoietin har visats i olika djurmodeller in vivo (normala och anemiska råttor, polycytemiska möss). Efter administrering av erytropoietin ökar erytrocyträkningen, hemoglobinvärden och retikulocytantal tillsammans med 59Fe -inkorporeringshastigheten. I tester in vitro (mus mjält cellkultur), ökad införlivande av 3H-tymidin i kärnbildade erytroidceller i mjälten observerades efter inkubation med erytropoietin.
Genom cellkulturer av mänsklig benmärg har det visats att erytropoietin specifikt stimulerar erytropoes utan att ändra leukopoies. Ingen cytotoxisk aktivitet av erytropoietin observerades på benmärgsceller.
På samma sätt som andra hematopoetiska tillväxtfaktorer har erytropoietin visats in vitro att ha stimulerande egenskaper hos humana endotelceller.
Vuxna patienter med nedsatt njurfunktion som ännu inte genomgår dialys
I 2 förlängda dosintervallstudier av erytropoietin (3 gånger i veckan, en gång i veckan, en gång varannan vecka och var fjärde vecka) upprätthöll vissa patienter med längre dosintervall inte tillräckliga hemoglobinnivåer och protokollkraven för avbrott som definierades av hemoglobinet värdet uppfylls (0% vid dosering en gång i veckan, 3,7% vid dosering varannan vecka och 3,3% i grupper var 4: e vecka).
Klinisk effekt och säkerhet
Tre placebokontrollerade studier involverade 721 cancerpatienter som fick platinafri kemoterapi, inklusive 389 med hematologiska maligniteter (221 med multipelt myelom, 144 med non-Hodgkins lymfom och 24 med andra hematologiska maligniteter) och 332 med solida tumörer (172 bröstceller, 64 gynekologiska , 23 lung-, 22 prostata-, 21 gastrointestinala och 30 andra). Två stora öppna studier omfattade 2 697 cancerpatienter som fick platinafri kemoterapi, inklusive 1 895 med solida tumörer (683 bröst, 260 lungor, 174 gynekologiska, 300 gastrointestinala och 478 andra) och 802 med hematologiska maligniteter.
I en prospektiv, randomiserad, dubbelblind, placebokontrollerad studie utförd på 375 anemiska patienter med olika icke-myeloida neoplasmer och som fick platinafri kemoterapi, en signifikant minskning av följdsjukdomarna i samband med anemi (såsom trötthet, asteni och minskad aktivitet ), mätt med följande bedömningsverktyg: den allmänna bedömningsskalan FACT-An (Functional Assessment of Cancer Therapy-Anemia), utmattningsskala FACT-An och Cancer Linear Analogue Scale (CLAS). Två andra randomiserade, placebokontrollerade studier med färre patienter visade inte en signifikant förbättring av livskvalitetsparametrar bedömda med EORTC-QLQ-C30-skala respektive CLAS.
Erytropoietin är en tillväxtfaktor som främst stimulerar produktionen av erytrocyter Erytropoietinreceptorer kan uttryckas på ytan av olika typer av tumörceller.
Överlevnad och tumörprogression analyserades i fem stora kontrollerade studier, som omfattade totalt 2833 patienter, inklusive fyra dubbelblinda, placebokontrollerade studier och en öppen studie. Dessa studier inkluderade patienter som fick kemoterapi (två studier) eller patientpopulationer där erytropoesstimulerande medel inte är indikerade: cancerpatienter med anemi som inte genomgår kemoterapi och patienter med huvud- och nackcancer., Som genomgår strålbehandling. I två studier, målhemoglobin koncentrationen var> 13 g / dL; i de återstående studierna var den 12-14 g / dL. I den öppna studien hittades ingen skillnad i total överlevnad mellan patienter behandlade med rekombinant humant erytropoietin kontra kontroller. I de fyra placebo- kontrollerade studier, riskförhållandet (riskförhållande) för total överlevnad var mellan 1,25 och 2,47, till förmån för kontroller. Jämfört med kontroller observerade dessa studier en statistiskt signifikant, konstant och oförklarlig ökning av dödligheten hos patienter med anemi i samband med flera vanliga maligniteter och behandlade med rekombinant humant erytropoietin. Studiernas överlevnadsresultat kunde inte på ett tillfredsställande sätt förklaras av skillnaderna i förekomst av trombos och associerade komplikationer hos patienter behandlade med rekombinant humant erytropoietin och hos kontrollpersoner.
En systematisk granskning genomfördes också på över 9 000 cancerpatienter som deltog i 57 kliniska prövningar. Metaanalys av övergripande överlevnadsdata gav ett uppskattat riskförhållande på 1,08 till förmån för kontroller (95% KI: 0,99, 1,18; 42 studier och 8 167 patienter). En ökad relativ risk för tromboemboliska händelser observerades hos patienter behandlade med rekombinant humant erytropoietin (RR 1,67, 95% KI: 1,35, 2,06, 35 studier och 6 769 patienter). Det finns en ökad risk för tromboemboliska händelser hos cancerpatienter som behandlas med rekombinant humant erytropoietin och en negativ effekt på den övergripande överlevnaden kan inte uteslutas. Det är inte känt i vilken utsträckning dessa data kan hänföras till administrering av rekombinant humant erytropoietin till cancerpatienter som får kemoterapi för att uppnå hemoglobinkoncentrationer under 13 g / dl, eftersom endast ett fåtal patienter med de beskrivna egenskaperna inkluderades i de granskade data.
En enda patientdataanalys utfördes också på över 13 900 cancerpatienter (kemoradio-, kemoradium- eller ingen terapi) som deltog i 53 kontrollerade kliniska prövningar med olika epoetiner. Total överlevnad genererade en riskförhållandepunktsuppskattning på 1,06 till fördel för kontroller (95% KI: 1,00, 1,12: 53 försök och 13 933 patienter) och för cancerpatienter som fick kemoterapi var det totala överlevnadsriskkvoten 1,04 (95% KI: 0,97, 1,11; 38 försök och 10 441 patienter). -analys stöder också en konsekvent och signifikant ökad relativ risk för tromboemboliska händelser hos cancerpatienter behandlade med rekombinant humant erytropoietin (se avsnitt 4.4).
I en randomiserad, dubbelblind, placebokontrollerad studie av 4038 icke-dialyserade CRF-patienter med typ 2-diabetes och hemoglobinvärden ≤ 11 g / dl, behandlades patienterna med antingen darbepoetin alfa för att uppnå hemoglobinnivåer på 13 g / dl eller placebo (se avsnitt 4.4). Studien uppfyllde inte några av de primära målen med att demonstrera att minska risken för relaterad dödlighet, kardiovaskulär morbiditet och utveckling av njursjukdom i slutstadiet (ESRD). Analyser av de enskilda komponenterna i de sammansatta slutpunkterna visade en HR (95% CI): död 1,05 (0,92, 1,21), stroke 1,92 (1,38, 2,68), hjärtsvikt (CHF) 0,89 (0,74, 1,08), hjärtinfarkt (MI) 0,96 (0,75, 1,23), sjukhusvistelse för myokardiskemi 0,84 ( 0,55, 1,27), ESRD 1,02 (0,87, 1,18).
Samlade analyser av post-hoc-data från kliniska prövningar med ESA utförda på patienter med CRF (på dialys, inte på dialys, med eller utan diabetes). Det fanns en trend mot ökande riskuppskattningar för dödlighet av alla orsaker och kardiovaskulära och cerebrovaskulära händelser i samband med de högsta kumulativa doserna av ESA oavsett diabetes eller dialysstatus (se avsnitt 4.2 och 4.4).
05.2 "Farmakokinetiska egenskaper -
Intravenös administreringssätt
Mätning av erytropoietin efter upprepad intravenös administrering visade en "halveringstid på cirka 4 timmar hos friska frivilliga och en" något längre halveringstid hos patienter med njurinsufficiens (cirka 5 timmar). En halveringstid på cirka 6 timmar har rapporterats hos barn.
Subkutan administreringssätt
Efter subkutan injektion är erytropoietinnivåerna i serum mycket lägre än intravenösa nivåer, långsamt ökar och toppar mellan 12 och 18 timmar efter administrering. Denna topp är alltid långt under det som nåddes intravenöst (cirka 1/20).
Det finns inga ackumuleringsfenomen: koncentrationerna förblir desamma, oavsett om de detekteras 24 timmar efter den första injektionen eller 24 timmar efter den sista injektionen.
Halveringstiden är svår att bedöma vid subkutan administrering och uppskattas till cirka 24 timmar. Biotillgängligheten för subkutan injicerbar erytropoietin är mycket lägre än det intravenösa läkemedlet: cirka 20%.
05.3 Prekliniska säkerhetsdata -
I vissa prekliniska toxikologiska studier på hundar och råttor, men inte på apor, har erytropoietinbehandling associerats med subklinisk benmärgsfibros (benmärgsfibros är en känd komplikation av kroniskt njursvikt hos människor och kan relateras till sekundär hyperparatyreoidism eller okända faktorer) . I en studie utförd på hemodialyspatienter som behandlats med erytropoietin i 3 år, ökade inte incidensen av benmärgsfibros jämfört med en motsvarande grupp kontrollpatienter på dialys men behandlades inte med erytropoietin.
Djurstudier har visat att erytropoietin minskar fostrets kroppsvikt, fördröjer ossifieringsprocessen och ökar fosterdödligheten när den ges i veckodoser ungefär 20 gånger de som rekommenderas för människor. Dessa förändringar tolkas som sekundära till den minskade ökningen av moderns kroppsvikt.
Erytropoietin visade ingen aktivitet i mutagenicitetstester på cellkulturer av bakterier och däggdjur och in vivo i ett musmikronukleustest. Långsiktiga cancerframkallande studier har inte utförts. Det finns motstridiga data i litteraturen om möjligheten att erytropoietin spelar en viktig roll i spridningen av cancerceller. Dessa data är baserade på erhållna resultat in vitro från humana tumörvävnadsprover; deras omfattning i klinisk miljö är dock oklar.
06.0 LÄKEMEDELSINFORMATION -
06.1 Hjälpämnen -
Dinatriumfosfatdihydrat
Natriummonobasiskt fosfatdihydrat
Natriumklorid
Kalciumkloriddihydrat
Polysorbat 20
Glycin
Leucine
Isoleucin
Threonine
Glutaminsyra
Fenylalanin
Vatten för injicerbara lösningar
Natriumhydroxid (för att justera pH)
Saltsyra (för att justera pH)
06.2 Inkompatibilitet "-
I avsaknad av inkompatibilitetsstudier får detta läkemedel inte blandas med andra produkter.
06.3 Giltighetstid "-
30 månader
06.4 Särskilda förvaringsanvisningar -
Förvaras i kylskåp (2 ° C - 8 ° C). Frys inte.
Förvara den förfyllda sprutan i ytterkartongen för att skydda läkemedlet från ljus.
Vid poliklinisk användning kan patienten ta ut produkten från kylskåpet och förvara den vid rumstemperatur (högst 25 ° C) under en period av högst 3 dagar.
06.5 Förpackningens innehåll och förpackningens innehåll -
0,3 ml lösning i förfylld spruta av typ I-glas med fast stålnål och PTFE-fodrad kolvpropp med eller utan nålskydd.
En förpackning innehåller 1 eller 6 förfyllda sprutor.
Alla förpackningsstorlekar kanske inte marknadsförs.
06.6 Anvisningar för användning och hantering -
Anvisningar för hantering av Retacrit:
1. Kontrollera att lösningen är klar, färglös och praktiskt taget fri från synliga partiklar efter att du har tagit bort en spruta från blistret.
2. Ta bort nålskyddet och pressa ut luften från nålen och sprutan genom att hålla sprutan upprätt och försiktigt skjuta upp kolven.
3. Sprutan är klar att användas.
Retacrit ska inte användas om något av följande inträffar:
• blåsan är öppen eller på annat sätt skadad;
• lösningen inte är färglös eller innehåller synliga partiklar i suspension;
• c "vätska har läckt ut från den förfyllda sprutan eller kondens är synlig inuti blistret som fortfarande är förseglat;
• läkemedlet frystes av misstag.
Detta läkemedel är endast avsett för engångsbruk.
Skaka inte.
Oanvänd medicin och avfall från detta läkemedel måste kasseras i enlighet med lokala föreskrifter.
07.0 INNEHAVARE AV "MARKNADSFÖRINGANDE GODKÄNNANDE" -
Hospira UK Limited
Horisont
Honey Lane
Hurley
Maidenhead
SL6 6RJ
Storbritannien
08.0 NUMMER FÖR FÖRSÄLJNINGSTILLSTÅND -
EU/1/07/431/001 förfylld spruta
EU/1/07/431/002 förfylld spruta
EU/1/07/431/026 förfylld spruta med nålskydd
EU/1/07/431/027 förfylld spruta med nålskydd
038381012
038381024
038381265
038381277
09.0 DATUM FÖR FÖRSTA GODKÄNNANDE ELLER FÖRNYELSE AV GODKÄNNANDE -
Datum för första godkännandet: 18 december 2007
Datum för senaste förnyelse: 15 november 2012
10.0 DATUM FÖR REVISION AV TEXTEN -
D.CCE september 2016
11.0 FÖR RADIOParma, FULLSTÄNDIGA DATA OM INTERN STRÅLNINGSDOSIMETRI -
12.0 FÖR RADIODROGAR, YTTERLIGARE DETALJERADE INSTRUKTIONER OM EXTEMPORÄR FÖRBEREDELSE OCH KVALITETSKONTROLL -