Allmänhet
Osteogenesis imperfecta är en medfödd genetisk sjukdom, som inte är kopplad till kön, som är ansvarig för en viss benbräcklighet och en tydlig tendens till frakturer.
Symtomen på osteogenesis imperfecta är många; i allmänhet består de av: benförsvagning, hög tendens till benfrakturer, förekomst av blå, grå eller lila okulära sklera, förekomst av bendeformiteter eller andra skelettförändringar, triangulärt ansikte, tandbräcklighet, etc. .
I allmänhet är följande avgörande för en korrekt diagnos av osteogenesis imperfecta: fysisk undersökning, medicinsk historia, medicinska bildtest, ett kollagatest av typ I och ett genetiskt test.
Tyvärr är de enda behandlingarna som är tillgängliga för patienter med osteogenes imperfecta för närvarande symtomatiska. Sjukdomen i fråga är faktiskt obotlig.
Vad är osteogenesis imperfecta?
Osteogenesis imperfecta är en genetisk sjukdom som gör den drabbade människans ben svagare och mer benägna att frakturer.
I verkligheten, med termen osteogenesis imperfecta, hänvisar läkare till en heterogen grupp av genetiska sjukdomar, kännetecknade av en viss grad av benbräcklighet. Det finns därför flera former (eller typer) av osteogenesis imperfecta, vissa mycket allvarligare än andra.
DET ÄR EN KONGENITAL SJUKDOM
Hos människor som drabbas av det är osteogenesis imperfecta en sjukdom som finns från födseln, och därför kan den definieras som en medfödd sjukdom.
ÄR DET SEX RELATERAT?
Osteogenesis imperfecta är inte en könsrelaterad genetisk sjukdom, såsom hemofili eller Klinefelters syndrom.
EPIDEMILOGI
Enligt viss statistisk forskning skulle förekomsten av osteogenesis imperfecta vara lika med ett fall var 15.000-20.000 födda. Detta innebär att var 15 000–20 000 nyfödda får en som drabbas av osteogenesis imperfecta.
Andra statistiska studier har också visat att osteogenesis imperfecta påverkar män och kvinnor lika mycket, och att det inte har någon preferens för en viss befolkning eller etnisk grupp.
Livslängd är en extremt variabel parameter, som beror på formen av osteogenesis imperfecta.
Orsaker
Osteogenesis imperfecta är nästan alltid resultatet av en kvalitativ och kvantitativ förändring av produktionen av typ I -kollagen.
Kollagen av typ I är viktigt för att stärka ben och för att upprätthålla friska bindväv som utgör brosk, senor, hud, okulär sclera etc.
Därför påverkar en förändring i produktionen av kollagen av typ I benets styrka och den goda hälsan hos de bindväv som finns i människokroppen.
VAD ÄNDRAR COLLAGEN -PRODUKTIONEN?
En genetisk sjukdom är ett tillstånd som uppstår på grund av en mutation av en eller flera gener som bildar cellulärt DNA.
När det gäller osteogenesis imperfecta är orsakerna till de senare nästan alltid att finna i mutationen av en eller båda generna COL1A1 (lokaliserad på kromosom 17) och COL1A2 (belägen på kromosom 7).
Under normala förhållanden reglerar COL1A1 och COL1A2 normal produktion av typ I -kollagen; i närvaro av mutationer i deras laddning misslyckas de i sin reglerande funktion.
Viktigt: vilka andra gener, om de är muterade, orsakar osteogenesis imperfecta?
Förutom mutationerna av COL1A1 och COL1A2 är mutationer i IFITM5, SERPINF1, CRTAP och LEPRE1 gener potentiella orsaker till osteogenes imperfecta.
De ovannämnda generna täcker funktioner som skiljer sig från COL1A1 och COL1A2 - därför kontrollerar de inte produktionen av kollagen av typ I - men de har fortfarande ett "inflytande på styrkan och motståndet hos benen i det mänskliga skelettet.
VILKEN GENETISK sjukdom är det?
Osteogenesis imperfecta är en autosomal genetisk sjukdom.
Termen autosomal, associerad med en genetisk sjukdom, indikerar att tillståndet i fråga beror på genetiska mutationer baserade på autosomala och icke-könskromosomer.
Läsarna påminns om att människan har en kromosomuppsättning med 23 par totala kromosomer, där 22 par är av autosomaltyp och endast ett par är av den sexuella typen. Paret av kromosomer av den sexuella typen påverkar kön individ.
Osteogenesis imperfecta efter mutationer i COL1A1, COL1A2 och IFITM5 har alla egenskaper hos en autosomal dominerande sjukdom.När det beror på mutationer i generna SERPINF1, CRTAP och LEPRE1, har det egenskaperna hos en autosomal recessiv sjukdom.
TYPER
För närvarande tror läkare att det finns 8 typer (eller former) av osteogenesis imperfecta. För att skilja de olika typerna bestämde de sig för att använda den romerska numreringen, för att vara exakt de första åtta romerska siffrorna.
Tabellen nedan visar de 8 formerna av osteogenesis imperfecta, mutationerna som orsakar dem och andra egenskaper.
Kille
Muterad gen
Typ av genetisk sjukdom
DE
COL1A1
Autosomal dominant
II
COL1A1 och COL1A2
Autosomal dominant
III
COL1A1 och COL1A2
Autosomal dominant
IV
COL1A1 och COL1A2
Autosomal dominant
V.
IFITM5
Autosomal dominant
DU
SERPINF1
Autosomal recessiv
VII
CRTAP
Autosomal recessiv
VIII
HARE 1
Autosomal recessiv
* NB: uppenbarligen är mutationerna i COL1A1 och COL1A2, som orsakar de fyra första formerna av osteogenesis imperfecta, genetiska förändringar med något olika egenskaper. Annars skulle det inte vara meningsfullt att skilja det ena från det andra.
Symtom, tecken och komplikationer
Alla typer av osteogenesis imperfecta är ansvariga för en försvagning av benen, så att personen som drabbas av sjukdomen har en särskild predisposition för frakturer. Graden av försvagning av benen varierar beroende på formen; för vissa av dessa är denna försvagning större än för andra.
Med detta sagt måste det påpekas att varje form av osteogenesis imperfecta har sin egen symptomatiska bild, som för vissa kanske påminner om den symptomatologiska bilden av andra former.
MÖJLIGA SYMPTOM OCH TECKEN
De möjliga symptomen och tecknen på osteogenesis imperfecta inkluderar:
- Förekomst av benmissbildningar;
- Förekomst av en kort och liten kropp (avsedd som bagageutrymme);
- Ledproblem (t.ex. lösa leder);
- Muskelsvaghet;
- Blå, lila eller grå okulär sclera;
- Triangulärt ansikte;
- Fat bröstet;
- Morfologiska avvikelser i ryggraden;
- Tandskörhet;
- Nedgång eller total förlust av hörsel;
- Andningsproblem
- Problem relaterade till frånvaro eller brist på typ 1 -kollagen.
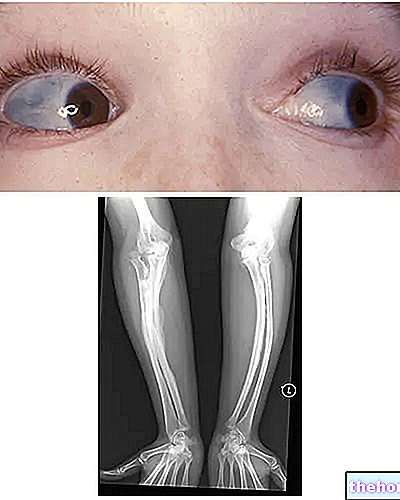
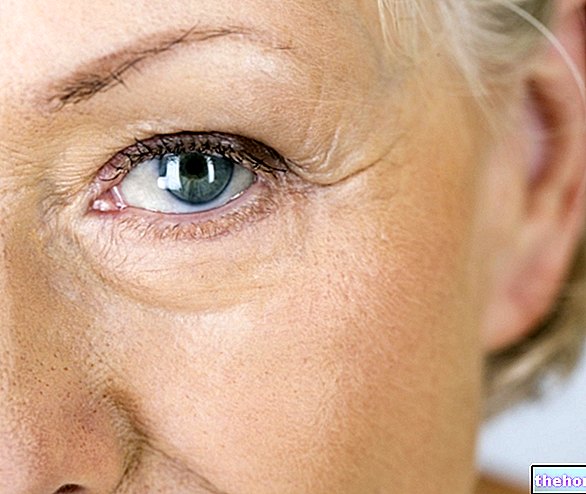
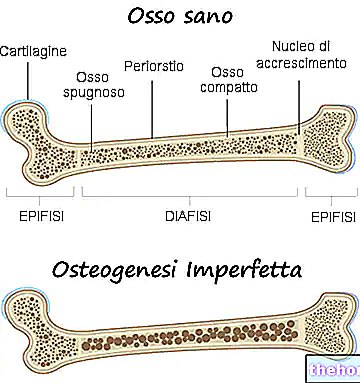
Osteogenesis Imperfecta: notera den blå färgen på sclerae och bendeformationerna som kännetecknar sjukdomen. Från wikipedia.org
VAD ÄR DE ALLVARLIGASTE FORMERNA FÖR IMPERFEKT OSTEOGENES?
Läkare klassificerar den symptomatologiska svårighetsgraden av de olika typerna av osteogenesis imperfecta på en skala av 3 grader, vilket är: den milda graden, den måttliga graden och den svåra graden.
Endast en form tillhör kategorin "mild grad": "typ I osteogenesis imperfecta"; 4 former av osteogenesis imperfecta tillhör kategorin "måttlig grad": IV, V och VI; slutligen tillhör kategorin "svår grad" 3 former: II, III, VII och VIII.
TYP I: FUNKTIONER
Den vanligaste och minst allvarliga formen av alla, typ I osteogenesis imperfecta har följande egenskaper:
- Det orsakar frakturer särskilt före puberteten;
- Det har "nästan inget inflytande på höjden, så patienterna brukar ha normal" höjd;
- Orsakar ledproblem och muskelsvaghet
- Den är ansvarig för blå, lila eller grå sclera;
- Det är orsaken till triangulära ansikte och ryggmärgsanomalier;
- Det orsakar nästan aldrig bendeformiteter. Om det provocerar dem är de minimala;
- Det kan orsaka tandskörhet och / eller hörselnedsättning (det senare inträffar vanligtvis i vuxen ålder);
- Det är associerat med förekomsten av typ I -kollagen som är normalt i kvalitet men onormalt i kvantitet (det är sämre än normalt).
TYP II: FUNKTIONER
Typ II osteogenesis imperfecta kännetecknas av:
- Dödsorsak vid födseln eller kort därefter. Andningsproblem orsakar nästan alltid död;
- Förekomst av betydande benbräcklighet och allvarliga bendeformiteter;
- Kort statur och underutvecklade lungor
- Blå, lila eller gråfärgad sclera;
- Förekomst av kvantitativa och kvalitativa avvikelser av kollagen av typ I.
TYPE III: FUNKTIONER
Typ III osteogenesis imperfecta har följande egenskaper:
- Även om det är mycket allvarligt, orsakar det inte ofta död under den nyfödda perioden;
- Det är associerat med "hög benbräcklighet;
- Den är ansvarig för kort statur, ledproblem, muskelsvaghet (särskilt i benen och armarna), fatbröstet, triangulärt ansikte och onormal krökning av ryggraden;
- Det beror på blå, lila eller grå sclera;
- Det kan orsaka andningsproblem, tandskörhet och hörselnedsättning;
- Det är ofta ansvarigt för bendeformiteter;
- Det är associerat med kvalitativa och kvantitativa abnormiteter av kollagen av typ I.
TYPE IV: FUNKTIONER
Typ IV osteogenes kännetecknas av:
- En grad av benbräcklighet mellan formerna II och III och form I;
- Kortare stativ än genomsnittet;
- Blå, lila eller gråfärgad sclera;
- Bendeformiteter hos mild / måttlig enhet, små avvikelser i ryggraden och fatbröstet;
- Triangulärt ansikte;
- Möjlig förekomst av tandbräcklighet och hörselnedsättning;
- Förekomst av typ I -kollagenavvikelser.
TYP V: FUNKTIONER
Typ V osteogenesis imperfecta liknar typ IV osteogenesis imperfecta på vissa sätt. Det har dock vissa särdrag, som är:
- Normal färgad sclera;
- Frånvaro av tandskörhet;
- Bildning av onormala beniga förhårdnader, under läkningsprocessen av benbrott;
- Förkalkning av det interosseösa membranet som ligger mellan radien och ulna. Detta försämrar rörligheten i underarmen.
TYP VI: FUNKTIONER
Också typ VI osteogenesis imperfecta liknar form IV. För att skilja den från den senare är några särdrag, inklusive de höga blodhalterna av alkaliskt fosfatas och förekomsten på vissa ben av lameller (beniga) som liknar fiskar.
TYP VII: FUNKTIONER
Symptomatiskt kan typ VII osteogenesis imperfecta likna typ IV under vissa omständigheter och typ II under andra omständigheter.
Egenskaperna hos denna allvarliga patologiska form inkluderar:
- Kort statur;
- Förekomsten av en extremt kort humerus (armben) och lårben (lårben);
- Den frekventa förekomsten av en höftdeformitet, känd som coxa vara.
TYPE VIII: EGENSKAPER
Typ VIII osteogenesis imperfecta påminner mycket om formerna II och III.
Bland dess särdrag utmärker sig följande: det allvarliga tillväxtunderskottet, den allvarliga skeletthypomineraliseringen och frånvaron (eller knapphet) av prolyl-3-hydroxylasenzymet.
Diagnos
I allmänhet börjar den diagnostiska process som patienter med en misstänkt form av osteogenesis imperfecta utsätts för med en noggrann fysisk undersökning och en noggrann medicinsk historia; sedan fortsätter det, med en "analys av patientens familjehistoria och med en serie diagnostiska avbildningstester (röntgen, CT-skanningar, etc.); slutligen slutar det med en kvantitativ och kvalitativ utvärdering av kollagen av typ I och med en genetiskt test.
Idag finns det möjlighet att diagnostisera osteogenesis imperfecta även i prenatalfasen genom att utsätta en gravid kvinna för ultraljud.
VIKTEN FÖR MÅLGRUNDLÄGGNINGEN OCH HISTORIEN
En läkarexpert i osteogenesis imperfecta kan mycket ofta diagnostisera den ovan nämnda sjukdomen endast med hjälp av en fysisk undersökning och anamnes. Detta innebär att dessa diagnostiska tester inte har någon försumbar betydelse.
UTVÄRDERING AV TYPE I COLLAGEN -PRODUKTION
Som regel är den kvalitativa och kvantitativa utvärderingen av kollagen av typ I ett mycket tillförlitligt test, eftersom de flesta fall av osteogenesis imperfecta, som sagt, kännetecknas av mutationer i generna som styr produktionen av typ 1 -kollagen.
För att bedöma kvantiteten och kvaliteten på typ I -kollagen som finns på cellnivå hos en individ kan läkare förlita sig på en hudbiopsi eller ett särskilt blodprov.
Båda dessa utvärderande tester är ganska komplexa och patienten (eller hans föräldrar) kan behöva vänta flera veckor för att få veta resultaten.
GENETISKT TEST
Genom ett genetiskt test som undersöker hela DNA: n för individen som undersöks kan läkare definitivt bestämma egenskaperna hos den närvarande genetiska mutationen.
I allmänhet förutses utförandet av ett genetiskt test på allt cellulärt DNA när utvärderingen av egenskaperna hos typ I -kollagen inte har gett de önskade resultaten, eller när det inte är en mutation i COL1A1 eller COL1A2 som orsakar "osteogenesis imperfecta.
PRENATAL DIAGNOS
Prenatal ultraljud är mycket användbart för att identifiera typ II och typ III osteogenesis imperfecta.
Terapi
Det finns för närvarande inget specifikt botemedel mot osteogenesis imperfecta. Med andra ord är personer med osteogenesis imperfecta avsedda att leva med ovannämnda tillstånd fram till döden, vilket ofta beror på konsekvenserna av själva sjukdomen.
Bristen på specifik behandling utesluter inte förekomsten av andra behandlingsformer. Faktum är att bland de terapeutiska möjligheterna för en patient med osteogenesis imperfecta ingår olika symptomatiska terapier; Med symptomatiska terapier menar vi behandlingar som kan lindra symtom, bromsa sjukdomsförloppet och förebygga (eller åtminstone skjuta upp) de allvarligaste konsekvenserna.
MÖJLIGA SYMPTOMATISKA BEHANDLINGAR
I listan över möjliga symtomatiska behandlingar för osteogenesis imperfecta sticker följande ut:
- Den kirurgiska insatsen, inuti de längsta benen (N.B: den mest benägna att frakturer), av naglar som ger större motståndskraft mot frakturer och missbildningar. Denna operation kallas roddning intramedullär;
- Konservativ eller kirurgisk behandling av frakturer och / eller bendeformiteter;
- Tandvård, för att skydda tändernas hälsa;
- Smärtlindrande behandlingar vid mycket smärtsamma multipla frakturer;
- Sjukgymnastik, för muskelförlängning och -förstärkning En elastisk och tonisk muskulaturapparat låter dig förhindra fall som kan leda till olika benfrakturer;
- Användning av hjälpmedel för rörelse, inklusive rullstolar, hängslen, kryckor etc.
FÖRDELAR MED RÖRELSE
För personer med osteogenesis imperfecta rekommenderar läkare konstant träning och rörelse i allmänhet, eftersom båda dessa aktiviteter bidrar till att stärka skelett- och muskelsystemet.
Bland de rekommenderade sporterna är: simning, eftersom det är en "låg påverkan fysisk aktivitet på skelettsystemet" och promenader.
FÖRDELAR FRÅN EN HÄLLSKA LIVSTIL
Att leva ett hälsosamt liv, undvika att röka, dricka överdriven alkohol, äta för mycket och dåligt, etc., har mer än diskreta hälsofördelar för patienter med osteogenesis imperfecta, eftersom det bromsar utvecklingen av sjukdomen och minskar benbräckligheten.
SYMPTOMATISKA BEHANDLINGAR I EXPERIMENTATIONSFASEN
För närvarande utvärderar läkare och forskare effektiviteten av vissa symtomatiska behandlingar, inklusive tillväxthormonbehandling och bisfosfonatbaserad intravenös och oral behandling.
För tillfället lovar resultaten från ovannämnda undersökningsbehandlingar gott för hela det medicinska samhället.
Prognos
Osteogenesis imperfecta är en sjukdom med en negativ prognos, eftersom den är obotlig, drastiskt äventyrar livskvaliteten och i vissa fall orsakar för tidig död hos det drabbade subjektet.
Det bör dock noteras att många individer med en mild form av osteogenesis imperfecta, även tack vare moderna symptomatiska behandlingar, kan leva ett trevligt och tillfredsställande liv.
Förebyggande
Tyvärr finns det för närvarande ingen förebyggande åtgärd mot osteogenesis imperfecta.