Allmänhet
Termen retinit pigmentosa (RP) identifierar en grupp genetiska sjukdomar som kännetecknas av progressiv retinal degeneration.

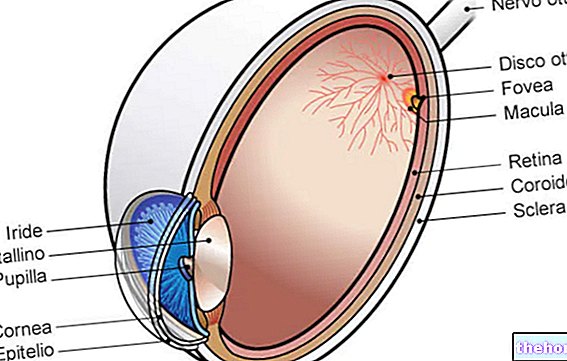
Retinitis pigmentosa är en retinal dystrofi som kännetecknas av gradvis förlust av fotoreceptorer och dysfunktion i pigmentepitelet, vilket innebär att näthinnan gradvis minskar sin förmåga att överföra visuell information till hjärnan via synnerven.
Den patologiska processen börjar med förändringar av retinalpigmentepitelet. När retinitis pigmentosa fortskrider blir det en gallring av blodkärlen som försörjer näthinnan, som genomgår atrofi. Vid undersökning av fundusen är de karakteristiska avlagringarna visuellt detekterbara. Retinalpigment ( därav namnet från sjukdomen). Atrofiska förändringar och skador kan också involvera synnerven och gradvis dör de ljuskänsliga cellerna i näthinnan.
Patienter som drabbats av retinitis pigmentosa upplever initialt synproblem särskilt i dåligt upplysta miljöer och klagar över en sammandragning av det perifera synfältet. Central vision sparas till de senare stadierna av sjukdomen, och det slutliga utfallet kan variera dramatiskt: många personer med retinitis pigmentosa behåller begränsad syn under hela sitt liv, medan andra tappar sikten helt.
Retinitis pigmentosa är en ärftlig sjukdom, främst orsakad av genetiska förändringar som överförs från en eller båda föräldrarna. Typen av genetisk defekt avgör vilka näthinneceller som är mest involverade i störningen och gör det möjligt att ur klinisk synvinkel skilja de olika tillstånden. Hittills har mer än 50 olika genetiska defekter som är inblandade i retinitis pigmentosa identifierats. Avvikelser kan överföras från föräldrar till avkommor genom ett av tre arvsmönster: autosomalt recessivt, autosomalt dominant eller heterosomalt recessivt (X-länkat eller X-kopplat).
Symtom
För ytterligare information: Retinitis Pigmentosa Symptom
Retinitis pigmentosa finns vanligtvis hos ungdomar och unga vuxna. Symtom uppträder ofta mellan 10 och 30 år, men diagnosen kan ställas i tidig barndom eller mycket senare i livet.
Tidiga symptom på retinitis pigmentosa kan innefatta:
- Svårt att se på natten (nattblindhet) eller i svagt ljus
- Långsam anpassning från synen i mörkret till den i ljuset, och vice versa;
- Förminskning av synfältet och förlust av perifer syn;
- Känslighet för ljus och bländning.
Vissa symptom beror på vilken typ av fotoreceptorer som är inblandade. Stavarna är ansvariga för svartvitt syn, medan kottarna gör att du kan skilja färger.
I de flesta fall av retinitis pigmentosa är stavarna inblandade först. Men i de snabbt utvecklande formerna kan kottar också påverkas i ett tidigt skede.
Stavar är koncentrerade i de yttre delarna av näthinnan och aktiveras av svagt ljus, så deras degeneration påverkar perifer och nattseende. Om kottar är inblandade är det möjligt att uppleva förlust av färguppfattning och central vision.
Övervägande av involverade fotoreceptorer bestäms av den speciella defekt som finns i patientens genetiska sammansättning.
Ofta är det första symptomet på retinitis pigmentosa nattblindhet (eller nattalopi). Vissa människor tycker att de behöver mer och mer tid för att anpassa sig till skillnader i ljus när de flyttar från ett väl upplyst område till ett mörkare. En typisk form av synförlust inducerar förminskning av perifer syn (tunnel- eller teleskopvision); detta mönster kallas ett ringskotom. Ibland kan detta fenomen saknas i de tidiga stadierna, men det märks när individen ofta snubblar över föremål eller är involverad i en trafikolycka. upplever svårigheter med läsning och detaljerat arbete som kräver koncentration på ett enda objekt, som att trä en tråd genom en nålsöga Många patienter rapporterar att de ser ljusglimtar (fotopsi), ofta beskrivna som små, flimrande och blinkande ljus.
Sjukdomsutvecklingen och graden av synförlust varierar från person till person. Vissa extrema fall kan utvecklas snabbt inom två decennier, andra en långsam kurs som aldrig leder till fullständig blindhet. Tidig debut finns i mer allvarliga former av retinitis pigmentosa, medan patienter med mildare tillstånd (t.ex. autosomaldominant) kan utveckla sjukdomen under sitt femte eller sjätte årtionde i livet.I familjer med X-linked retinitis pigmentosa drabbas män oftare än kvinnor och allvarligare; honor, å andra sidan, överför den genetiska egenskapen (de bär den förändrade genen på X -kromosomen) och uppvisar symtom på sjukdomen mindre ofta.
Komplikationer
Retinitis pigmentosa kommer att fortsätta att utvecklas, om än långsamt. Fullständig blindhet är dock sällsynt, men signifikant minskning av perifer och central syn kan uppstå.
Patienter med retinit pigmentosa utvecklar ofta svullnad i näthinnan (makulaödem) eller grå starr i tidig ålder. Dessa komplikationer kan behandlas om de stör synen.
Relaterade sjukdomar
Vanligtvis har en patient med retinit pigmentosa inga andra störningar och i det här fallet talar vi om "icke-syndromisk" eller enkel retinit pigmentosa. Flera syndrom delar dock några kliniska symptom med denna ögonsjukdom; det vanligaste är Usher syndrom, som drabbar cirka 10-30% av alla patienter med retinitis pigmentosa och är associerad med samtidig medfödd eller progressiv hörselnedsättning. I Lebers medfödda amauros kan barn dock bli blinda eller nästan blinda under de första sex månaderna av livet. Andra sjukdomar relaterade till retinitis pigmentosa inkluderar Bardet-Biedl syndrom och Refsums sjukdom.
Orsaker
Sjukdomen kan orsakas av ett antal genetiska defekter: i själva verket finns det flera gener som, om de påverkas av förändringen, kan orsaka retinitis pigmentosa fenotyp. Dessa kodar normalt för proteiner som är involverade i transduktionskaskaden som möjliggör syn, faktorer celltranskription (som skickar felaktiga meddelanden till näthinneceller) eller för element som utgör strukturen hos fotoreceptorer. Ärftliga genmutationer finns i celler från befruktningstillfället; vanliga avvikelser inkluderar de hos RP1-gener (i retinitis pigmentosa-1, autosomal dominant) , RHO (RP4, autosomal dominant) och RDS (RP7, autosomal dominant). Icke-ärftliga orsaker till retinitis pigmentosa är sällsynta, men möjligheten att hitta ett isolerat fall (spontan mutation), där det inte finns en familjehistoria av sjukdomen.