Vad är fenylketonuri
där fenylketonuri (P.K.U.) det är en autosomal recessiv ärftlig metabolisk sjukdom som drabbar 1 av 10 000 individer och verkar förekomma mer i homozygositet än i heterozygoter.
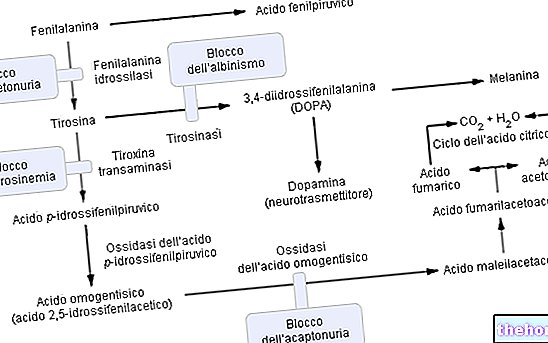
Tillhör gruppen hyperfenylalaninemi äventyrar fenylketonuri signifikant metabolismen av fenylalanin och i synnerhet dess konvertering till tyrosin; fenylketonuri erkänns av de förhöjda urinhalterna av fenylalanin och vissa derivat (fenylpyruvat, fenylacetat, fenylaktat och fenylacetylglutamin).
Den allvarligaste komplikationen av fenylketonuri är mental försening.
Fenylalanin, tyrosin och derivat
Fenylalanin är en essentiell aminosyra och utgör majoriteten av kostproteiner; det kan omvandlas av enzymet fenylalaninhydroxylas i tyrosin (genom tillsats av en hydroxylgrupp -OH). I sin tur är tyrosin en föregångare aminosyra för syntesen av:
- L-DOPA (dopaminsyntesintermediär)
- Epinefrin
- Noradrenalin (alla signalsubstanser).

Mekanism för fenylketonuri (P.K.U.)
Som förväntat är fenylketonuri, på grund av en eller flera (6 totalt) kromosomala mutationer, uttrycket (därmed den metaboliska aktiviteten) av fenylalaninhydroxylas praktiskt taget noll. Dessa förändringar kan vara av olika slag (från "missense" -ändringar till "splitsnings" -defekter eller till och med "partiella raderingar") men det som spelar roll är att på grund av denna enzymatiska ineffektivitet är fenylalaninnivåerna i blodet (som normalt är 1 mg / 100 ml) i DOMINANT fenylketonuri de når lätt mängder till och med 50 gånger högre.
Funktion av fenylalaninhydroxylasenzymet: För att producera tyrosin (+ dihydrobiopterin) kräver fenylalaninhydroxylas: fenylalanin, syre och tetrahydrobiopterin (en reducerad pteridin som fungerar som en coofactor); reaktionen är också reversibel och dihydrobiopterin kan rekonverteras (tack vare enzymet dihydropterinreduktas) i tetrahydrobiopterin.
Komplikationer
Fenylketonuri kan ge upphov till mer eller mindre allvarliga komplikationer baserat på svårighetsgraden av den patologiska manifestationen och diagnosens aktualitet; som en ärftlig patologi, skiljer sig fenylketonuri i:
- Dominant, därför kännetecknad av HELT inaktivitet av fenylalaninhydroxylasenzymet
- Recessiv, där endast 30% av det totala enzymatiska arvet är aktivt.
Komplikationerna av fenylketonuri kan hänföras till och direkt stå i proportion till den metaboliska ackumuleringen av fenylalanin, dess derivat och den minskade syntesen av tyrosin.I patologi filtreras överskottet av fenylalanin relativt effektivt av njurarna som endast delvis reabsorberar det och eliminerar det med urin ; Emellertid bestämmer persistensen av nivåerna av hyperfenylalaninemi en metabolisk reaktion av molekylär konvertering i fenylpyruvinsyra och / eller andra derivat lättare att dränera (fenylpyruvat, fenylacetat, fenylaktat).
Vad som komplicerar fenylketonuri är toxiciteten av fenylalanin, fenylpyruvinsyra och dess derivat mot centrala nervsystemet (CNS). Deras överdriven närvaro i hjärnans utveckling avgör obevekligt en form av mental retardation.
OBS. Plasmakoncentrationerna av de andra aminosyrorna är något reducerade, troligen på grund av återkoppling på tarmabsorption eller renal tubulär reabsorption.
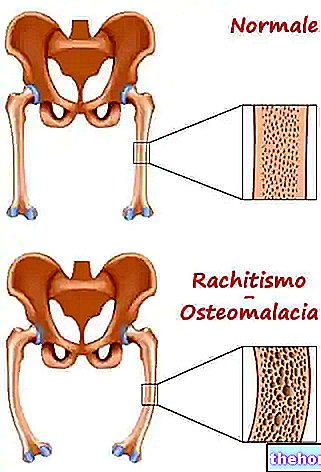
Hjärnskador, som en allvarlig komplikation av fenylketonuri, orsakas av subtraktion av andra väsentliga aminosyror vid proteosyntes, särskilt i bildandet av polyribosomer, myelin, noradrenalin och serotonin. Fenylketonuri - syns inte direkt efter födseln men efter några år - om det inte behandlas kräver sjukhusvistelse av barnet och är helt irreversibel.
Avancerad fenylketonuri kan också vara tydligt synligt för blotta ögat; de höga koncentrationerna av fenylalanin, som hämmar enzymet tyrosinas, försämrar signifikant syntesen av melanin genom att minska hud- och hårpigmentering; dessutom ger ackumulering av fenylacetat i hår och hud fenylketonurika en stark och obehaglig "muslukt".